Уникальные лекарственные средства что это
Автор: К.С.ДАВЫДОВА, филиал «Клиническая фармакология» НЦ БМТ РАМН
Большая часть лекарственных средств ( ЛС ) на современном фармрынке являются воспроизведенными (генерическими) препаратами. Согласно данным розничного аудита (IMS Health и DSM Group) доля дженериков в настоящее время составляет от 77 до 88% в натуральном выражении [5] (существуют данные и про долю в 95% [9], однако ее достоверность вызывает сомнения), при этом согласно прогнозам эта доля будет неуклонно расти. По объему генерического сектора Россия занимает 3 место в мире после Китая и Индии [2,3]. В то же время структура рынка стран большой семерки формируется следующим образом: в США — 12% дженериков, в Японии — 30%, в Германии — 35%, во Франции — 50%, в Англии — 55%, в Италии — 60%, в Канаде — 64% (рис. 1) [8].
Среди препаратов, которые ежегодно регистрируются в России, отмечается значительно большее количество дженериков, чем оригинальных препаратов. Отдельные оригинальные препараты имеют значительное количество воспроизведенных ЛС. Так, оригинальное лекарственное средство Вольтарен (действующее вещество – диклофенак натрия) сегодня имеет 207 дженериков, зарегистрированных к медицинскому применению. Также в РФ зарегистрировано около 150 генерических ЛС эналаприла, около 100 — нифедипина, атенолола, ципрофлоксацина и порядка 50 – нитроглицерина, аспирина и парацетамола (причем количество комбинированных генерических последних двух ЛС превышает 300) [1,4]. В ряде стран с развитой системой контроля качества, эффективности и безопасности ЛС, число дженериков инновационного препарата в большинстве случаев не превышает 4-5 [10,19].
Генерические ЛС выводятся на рынок после истечения срока патентной защиты. Они должны полностью соответствовать оригинальному продукту по составу действующих веществ (вспомогательные вещества могут быть иными) и лекарственной форме, соответствовать фармакопейным требованиям, быть произведенными в условиях GMP. В Федеральном законе о лекарственных средствах №86-ФЗ от 1998 г. дается определение воспроизведенных ЛС: «воспроизведенные лекарственные средства — лекарственные средства, поступившие в обращение после истечения срока действия исключительных патентных прав на оригинальные лекарственные средства». Однако такое определение не характеризует дженерик как копию или аналог инновационного препарата. В новом Федеральном законе «Об обращении лекарственных средств» №61-ФЗ от 2010 г. смысл термина раскрывается более полно, согласно современным международным рекомендациям: «воспроизведенное лекарственное средство — лекарственное средство, содержащее такую же фармацевтическую субстанцию или комбинацию таких же фармацевтических субстанций в такой же лекарственной форме, что и оригинальное лекарственное средство, и поступившее в обращение после поступления в обращение оригинального лекарственного средства».
Воспроизведенные ЛС имеют ряд равнозначных общеупотребляемых синонимов – «генерики», «дженерики», «генерические лекарственные средства», «многоисточниковые (мультиисточниковые) лекарственные средства» [8,9], однако согласно Федеральному закону № 61-ФЗ именно термин «воспроизведенные лекарственные средства» должен применяться в первую очередь. В то же время Всемирная организация здравоохранения в качестве основного понятия таких ЛС рекомендует употреблять термин «многоисточниковые лекарственные средства» (multisource drugs) [20]. Оригинальное (инновационное) лекарственное средство – это ЛС, которое было впервые зарегистрировано на основе полной документации в отношении его качества, безопасности и эффективности, защищенное патентом на срок до 20 лет [11].
Основными характеристиками оригинального ЛС являются: длительность разработки (10-15 лет) на основании отбора действующего вещества из значительного количества молекул; фармакологический эффект, токсичность, мутагенность и тератогенность которого проверены в доклинических исследованиях на животных; прохождение всех фаз клинических исследований в соответствии со стандартами GСP.
Несмотря на то что инновационное и воспроизведенное ЛС содержат одно и то же действующее вещество в одинаковой дозировке и лекарственной форме, эффективность и безопасность генерических препаратов может существенно различаться. Основными причинами таких различий могут быть фармацевтическая технология производства лекарственного препарата, вспомогательные вещества (неактивные ингредиенты, наполнители, консерванты, красители и др.), их природа и количество, полиморфизм, солевая форма, упаковка препарата, условия его хранения и транспортировки. Из-за этих отличий эффективность генерических препаратов и выраженность их побочных эффектов может сильно варьировать.
При этом следует отметить, что стоимость воспроизведенного ЛС ниже, чем оригинального, что определяется рядом причин. Для этого необходимо рассмотреть, из чего складывается стоимость оригинальных ЛС и дженериков. 80% стоимости оригинального ЛС составляет стоимость исследований эффективности и безопасности препарата, а 20% стоимости – это стоимость синтеза лекарственного вещества. Процесс создания оригинального ЛС является очень длительным и дорогостоящим. Сначала создается молекула, потом она оценивается в исследованиях на клетках и тканях, затем на животных. После этого следуют три этапа клинических исследований на здоровых добровольцах и пациентах. После завершения клинических исследований ЛС проходит регистрацию. Исследование оригинального ЛС продолжается и после регистрации. С соблюдением правил GCP проводятся и пострегистрационные исследования [23].
Известно, что только 1 из 5 000 молекул доходит до рынка в виде ЛС. Этот путь продолжается 12-15 лет, его стоимость составляет от 800 млн. до 1 млрд. долл. Прибыльными являются только 1-2 из вновь созданных ЛС [8]. Объяснением более низкой стоимости генерических ЛС являются: отсутствие клинических исследований; отсутствие масштабных доклинических исследований фармакологической активности, поисковых исследований; отсутствие изучения полного профиля безопасности.
Несмотря на широкое использование понятия эквивалентность, «эквивалентности» дженериков как термина не существует. Всемирная организация здравоохранения предлагает применять термин «взаимозаменяемость» (interchangeability) воспроизведенных лекарственных препаратов [23]. Взаимозаменяемое генерическое ЛС – это терапевтически эквивалентное генерическое ЛС, которым можно заменить препарат сравнения в клинической практике [22]. Видов «эквивалентности» воспроизведенных ЛС выделяют несколько – терапевтическая, фармацевтическая, биологическая, а также т.н. «эквивалентность in vitro» (in vitro equivalence), введенная в употребление в документе «WHO Technical Report Series 937. WHO Expert Committee on Specifications for Pharmaceutical Preparations (2006). Annex 7. Multisource (generic) pharmaceutical products: guidelines on registration requirements to establish interchangeability» [23].
Терапевтически эквивалентными лекарственные препараты могут считаться только в том случае, если они фармацевтически эквивалентны и можно ожидать, что они будут иметь одинаковый клинический эффект и одинаковый профиль безопасности при использовании пациентами в соответствии с указаниями инструкции по применению [7,22]. Терапевтическая эквивалентность означает, что два препарата обеспечивают одинаковый терапевтический эффект и безопасность. Терапевтически эквивалентные лекарственные препараты должны отвечать следующим требованиям: иметь доказанную эффективность и безопасность; быть фармацевтически эквивалентными; быть биоэквивалентными; иметь сходные инструкции по применению; производиться в условиях стандарта GMP [6]. Доказанную клиническую эффективность и безопасность устанавливают на основании клинических исследований.
ЛС считаются фармацевтически эквивалентными, если они содержат одни и те же действующие вещества в одинаковом количестве и в одинаковой лекарственной форме и отвечают требованиям одних и тех же или сходных стандартов [1]. То есть фармацевтическая эквивалентность – это полное соответствие состава и лекарственной формы препаратов. Для некоторых лекарственных форм фармацевтическая эквивалентность будет обеспечивать терапевтическую эквивалентность и, соответственно, взаимозаменяемость двух препаратов (препараты для местного применения, порошки для изготовления растворов, инъекционные растворы и некоторые другие) [23].
Отдельно стоит выделить фармацевтическую альтернативность ЛС. Лекарственные средства являются фармацевтически альтернативными, если они содержат одинаковое количество одной и той же активной субстанции (субстанций), но различаются по лекарственной форме (например, таблетки и капсулы) и/или по химической форме (различные соли, эфиры) [1].
Оценка биоэквивалентности ЛС является основным видом медико-биологического контроля воспроизведенных (генерических) ЛС, не отличающихся лекарственной формой и содержанием действующих веществ от соответствующих оригинальных ЛС. Биоэквивалентность ЛС обозначает их одинаковую биодоступность. Под биодоступностью понимают количество неизмененного действующего вещества, достигающего системного кровотока (степень всасывания) относительно исходной дозы ЛС. Исследования биоэквивалентности позволяют сделать обоснованные заключения о качестве сравниваемых препаратов по относительно меньшему объему первичной информации и в более сжатые сроки, чем при проведении клинических исследований [17,18,21].
В некоторых международных руководствах введено понятие регуляторной процедуры «биовейвер», в соответствии с которой определение взаимозаменяемости генерических ЛС проводится на основании оценки их биофармацевтических свойств и эквивалентности in vitro (изучение сравнительной кинетики растворения) либо другими методами in vitro в качестве альтернативы исследованиям биоэквивалентности in vivo при их государственной регистрации [16].
Самое главное, к чему надо стремиться, — дженерики, как и инновационные (оригинальные) препараты, должны отвечать требованиям, предъявляемым в рамках Общего (или единого) технического документа (CTD): эффективность, безопасность, качество 13, поэтому весь объем исследований должен быть достаточным для подтверждения данных требований.
Литература
1. Арзамасцев А.П., Дорофеев В.Л. Эквивалентность воспроизведенных лекарственных средств: фармацевтические аспекты. // Ведомости НЦЭСМП. – М., 2007. – №1. – С. 27-35.
2. Баула О.Ю. Современные регуляторные требования к исследованиям и регистрации генерических лекарственных средств. – М., «Фармсодружество», 2007.
3. Белоусов Ю.Б. Дженерики – мифы и реалии. «Ремедиум». – 2003. — № 7–8. — С. 4–9.
4. Верткин А.Л., О.Б.Талибов. Генерики и эквивалентность – что стоит за терминами. Неотложная терапия. — 2004; — № 1–2. – С.16–17.
5. Новикова Н.Н. // Фармацевтический Вестник. – М., 2008. – №4. – С. 4.
6. Рудык Ю.С. К вопросу о терапевтической эквивалентности лекарственных средств // Рациональная фармакотерапия. – Киев, 2007. — №2. – С. 40-48.
7. Семинар-тренинг ВОЗ по проведению теста растворения, взаимозаменяемости лекарственных средств и системе биофармацевтической классификации. // Аптека, — Киев, 2007. – № 31. – С. 10-17.
8. Талибов О.Б. Генерики и эквивалентность лекарственных препаратов. // Медицинская газета «Здоровье Украины». – Киев, 2008. – №5. – С. 12-16.
9. Тарловская Е.И. Генерики и оригинальные препараты: взгляд практического врача. // Российский Медицинский Журнал. – М., 2008, – т. 16. – №5. – С. 30 – 35.
10. Чумак В.Т. Оборот лекарственных средств в Украине. Проблемы и перспективы. Материалы І Международной конференции «Клинические испытания лекарственных средств в Украине». — Киев, 2006.
11. Directive 2004/27/EC of the European Parliament and of the Council, Art. 10.1. – 2004.
12. ICH Harmonised Tripartite Guideline: The Common Technical Document for the Registration of Pharmaceuticals for Human Use: Quality M4Q (R1). – Geneva: ICH, 2002.
13. ICH Harmonised Tripartite Guideline: The Common Technical Document for the Registration of Pharmaceuticals for Human Use: Safety. M4S (R2). – Geneva: ICH, 2002.
14. ICH Harmonised Tripartite Guideline: The Common Technical Document for the Registration of Pharmaceuticals for Human Use: Efficacy. M4E (R1). – Geneva: ICH, 2002.
15. Interchangeability of multisource drug products containing highly variable drugs. WHO/FIP Training Workshop on Dissolution, Pharmaceutical Product Interchangeability and Biopharmaceuticals Classification System (BCS) — Kiev, 2007.
16. Yu G., Amidon J. Polli. Biopharmaceutics Classification System: The Scientific Basis for Biowaiver Extensions. // Pharmaceutical Research. – 2002. – Vol. 19, №. 7.
17. Chen M., Lesko L. Individual bioequivalence revisited. // Clin Pharmacokinet – 2001. – № 40: – pp. 701-706.
18. Chen M., Shah V., Patnaik R. Bioavailability and Bioequivalence: An FDA Regulatory Overview. // Pharmaceutical Research. – 2001. – Vol. 18, №. 12.
19. Laroche M., Merle L. Generic and brand-name drugs. Are different criteria sufficiently taken into account before granting market authorisation? // Acta Clin Belg Suppl. – 2006. – № 1: – pp. 48-50.
20. Multisource (Generic) Pharmaceutical Products: Guidelines on Registration Requirements to Establish Interchangeability. — WHO Technical Report Series, № 937. – WHO, 2006.
21. Quality of bioequivalence data. WHO workshop on assessment of bioequivalence data submitted to regulatory authorities – Kiev, 2009.
22. WHO Technical Report Series 937. WHO Expert Committee on Specifications for Pharmaceutical Preparations. – WHO, 2006.
23. WHO Technical Report Series 937, annex 7 « Multisource (generic) pharmaceutical products: guidelines on registration requirements to establish interchangeability». WHO Expert Committee on Specifications for Pharmaceutical Preparations. – WHO, 2006.
Рисунок — в приложении
Файл: Загрузить (85 кбайт)
Источник
Самое дорогостоящее лекарство в мире
Автор
Редакторы
Статья на конкурс «био/мол/текст»: Не так давно, в мае 2019 года, произошло знаковое событие в сфере лечения генетических заболеваний: Управление по санитарному надзору за качеством продуктов и медикаментов США (FDA) одобрило препарат Zolgensma («Золгенсма», или онасемноген абепарвовек). Это лекарственное средство предназначено для генотерапевтического лечения спинально-мышечной атрофии (СМА). Сегодня «Золгенсма» является самым дорогим лекарственным препаратом в мире.

Конкурс «био/мол/текст»-2019
Эта работа опубликована в номинации «Свободная тема» конкурса «био/мол/текст»-2019.

Генеральный спонсор конкурса и партнер номинации «Сколтех» — Центр наук о жизни Сколтеха.
Спонсор конкурса — компания «Диаэм»: крупнейший поставщик оборудования, реагентов и расходных материалов для биологических исследований и производств.

Спонсором приза зрительских симпатий выступила компания BioVitrum.
Что такое СМА?
Спинально-мышечной атрофией, или СМА, называют смертельно опасное нейродегенеративное заболевание, в процессе развития которого у пациента происходит постепенная атрофия скелетной мускулатуры. В результате человек теряет или так и не приобретает способности ходить, самостоятельно стоять, сидеть без поддержки. Со временем возникает сколиоз и другие ортопедические проблемы. Также СМА-пациенты, если они не получают должного ухода и лечения, постепенно утрачивают способность самостоятельно дышать, глотать, кашлять. Пациенты с первым, самым тяжелым, типом СМА еще несколько лет назад, как правило, не доживали и до двух лет [1].
СМА возникает из-за потери участка хромосомы или точечной мутации гена SMN1, расположенного в пятой хромосоме. В результате этого нарушается синтез SMN-белка, недостаток которого приводит к гибели моторных нейронов и атрофии скелетной мускулатуры [2].
Для того чтобы болезнь проявилась, носителем рецессивной мутации в гене SMN1 должны быть оба родителя. Примерно каждый 40-й житель Земли является таким носителем.
Подробнее о причинах возникновения, диагностике, течении спинально-мышечной атрофии читайте в статье «Надежда для СМАйликов» [3].
Терапия спинально-мышечной атрофии до появления «Золгенсмы»
До недавнего времени методы лечения СМА сводились к поддерживающей терапии. Больным рекомендовали специальное питание, витамины, умеренные физические нагрузки, при необходимости — хирургическое вмешательство, искусственная вентиляция легких. К сожалению, до сих пор значительная часть СМА-пациентов получает лишь такое лечение.

Рисунок 1. «Спинраза» — первый препарат, одобренный для лечения СМА
С 2016 года сначала в США, а затем и в Европе для лечения спинально-мышечной атрофии стали применять препарат «Спинраза» (нусинерсен) [4]. Он позволяет существенно увеличить продукцию полноценного SMN-белка, что ведет к сглаживанию симптомов заболевания. Терапия тем эффективнее, чем меньше возраст пациента.
Стоимость препарата составляет несколько сот тысяч долларов в год, поэтому его закупка осуществляется не за счет пациента. Одна за другой страны разных континентов одобрили препарат и стали применять для спасения жизней своих сограждан. В некоторых государствах этот процесс сильно затянулся из-за бюрократических проволочек и нехватки финансирования.
В РФ «Спинразу» Минздрав одобрил в начале 2019. В свою очередь компания «Биоген», производитель «Спинразы», в апреле 2019 г. объявила об открытии в России «Программы расширенного доступа» для лечения СМА нусинерсеном. Благодаря этой программе доступ к препарату получили 40 детей из России, страдающих СМА I типа [5].
В августе «Спинраза» была включена в Государственный реестр лекарственных средств РФ. Однако охват больных все еще слишком мал. В Российской Федерации зарегистрировано около 800 СМА-пациентов, и далеко не все они получают инъекции «Спинразы».
Отличия «Золгенсмы» от «Спинразы»
После появления на фармацевтическом рынке «Спинразы» все ждали выхода принципиально нового препарата для лечения СМА, основанного на генотерапевтическом подходе. Лидером в данной разработке оказалась компания «Новартис» (Novartis), которая в 2018 году купила компанию «Авексис» (AveXis) за 8,7 млрд долларов, а в 2019 вышла на рынок c препаратом «Золгенсма» (Zolgensma, он же AVXS-101, или онасемноген абепарвовек) [6].
Рисунок 2. Логотип препарата «Золгенсма»
Чем же «Золгенсма» принципиально отличается от препарата «Спинраза»? Самое важное различие заключается в механизме действия: «Спинраза» исправляет дефект сплайсинга матричной РНК гена SMN2, но она никак не затрагивает ген SMN1, мутации в котором и являются основной причиной развития спинально-мышечной атрофии.
Действие же препарата «Золгенсма» направлено именно на ген SMN1. Благодаря использованию этого лекарственного средства, мутировавший или отсутствующий ген SMN1 замещается функционально полноценным геном [1].
Происходит это следующим образом: препарат содержит функционально полноценный ген SMN1, который находится внутри вектора. Задача вектора — быстро доставить его в мотонейроны тела (рис. 3).
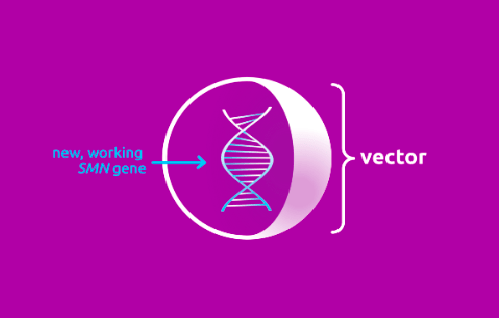
Рисунок 3. Условное изображение вектора, входящего в состав «Золгенсмы»
Для создания вектора использовали аденоассоциированный вирус (adeno-associative virus 9, или AAV9). Это представитель семейства парвовирусов, который способен инфицировать клетки человека и других приматов, но при этом не является патогенным. Все это делает AAV9 отличным генетическим вектором. Собственный генетический материал вируса удалили и вместо него поместили функционально полноценный ген SMN1 (рис. 4).
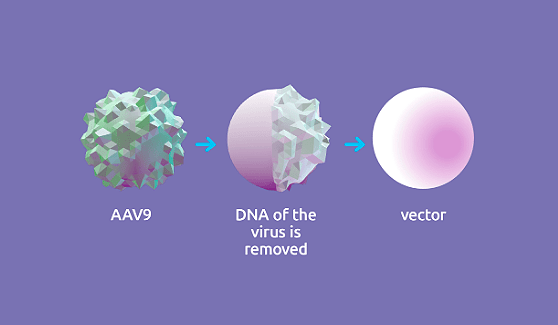
Рисунок 4. Условная схема механизма создания вектора
После того, как ген прибывает в нужную локацию, вектор разрушается и выводится из организма.
СМА-пациенту необходима всего одна инфузия препарата «Золгенсма» в течение жизни, в то время как лечение «Спинразой» требует нескольких доз в год. Отсюда и стоимость «Золгенсмы»: 2 125 000 долл. США. Такая ценовая политика компании-производителя делает данный препарат самым дорогим лекарственным средством на сегодняшний день. Для сравнения, стоимость все той же «Спинразы» составляет 125 тыс. долларов за одну дозу. При этом в первый год лечения нужно шесть инфузий, в последующие периоды — три инфузии ежегодно.
Насколько оправдана такая цена препарата и какова же его себестоимость? «Новартис» не афиширует информацию относительно себестоимости «Золгенсмы», поэтому эксперты оценивают стоимость препарата по двум показателям: качество жизни пациента с учетом прожитых лет (QALY) и добавленные годы жизни (LYG). По данным Института клинико-экономической экспертизы (Institute for Clinical and Economic Review, ICER), исходя из показателя QALY стоимость «Золгенсмы» должна быть в пределах 1,1–1,9 миллионов долл. США; исходя из показателя LYG — 1,2–2,1 миллиона долл. США. Таким образом, можно сказать, что стоимость «Золгенсмы» завышена по отношению к оценкам экспертов [7].
При формировании своей ценовой политики компания «Новартис» отталкивалась от стоимости препарата «Спинраза». По задумке производителя в течение десяти лет на лечение СМА-пациента «Спинразой» необходимо будет потратить более 4 млн долларов, в то время как одна инфузия «Золгенсмы» стоит 2 млн 125 тысяч. Таким образом, в долгосрочной перспективе второй вариант более выгоден [8], [9].
Компания «Новартис» ожидала, что «Золгенсма» станет «блокбастером», то есть принесет более 1 млрд долларов за первый год продаж. Однако скандал, который возник вокруг «Золгенсмы», может не дать осуществиться этим планам. Летом 2019 г. компания «Новартис» сама сообщила FDA о манипуляции с данными при проведении тестирования препарата на животных. Если бы эти данные были известны FDA в мае, то разрешение на использование препарата «Золгенсма» «Новартис» получила бы позже, но сейчас принято решение не отзывать препарат [10].
Сумму в два с лишним миллиона долларов не в состоянии заплатить большинство СМА-семей, поэтому предполагается, что пациенты будут обеспечиваться жизненно важным лечением благодаря государственной поддержке или за счет страховых компаний. Кроме того, производитель «Золгенсмы» предоставляет пятилетнюю рассрочку на оплату генной терапии и дает возможность пациенту не выплачивать оставшуюся сумму, если препарат перестанет действовать [8]. Сегодня препарат «Золгенсма» доступен только для жителей США, так как FDA — это единственная организация, которая его одобрила. Также есть ограничения по возрасту и тяжести заболевания: пока препарат применяется только для пациентов до двух лет с первым типом СМА. В дальнейшем производитель планирует использовать препарат и для других групп людей, страдающих спинально-мышечной атрофией.
Важно также отметить, что «Золгенсма» вводится внутривенно. «Спинраза» же должна попасть в спинномозговую жидкость пациента, что создает ряд дополнительных проблем и рисков.
Побочные эффекты «Золгенсмы»
Кроме высокой стоимости, у «Золгенсмы» есть и другие серьезные недостатки. Возможными побочными эффектами препарата являются:
- рвота;
- повышение уровня аминотрансфераз;
- тромбоцитопения;
- нарушение функций печени, вплоть до острого тяжелого поражения.
Не рекомендуется использование «Золгенсмы» у недоношенных детей до достижения ими полного гестационного возраста.
По данным компании-производителя на момент регистрации «Золгенсма» в рамках клинических исследований была применена для терапии 44 детей в возрасте от 0,3 до 7,9 месяцев с массой тела от 3 до 8,4 кг. Такая небольшая выборка объясняется тем, что СМА является редким заболеванием, поэтому набрать большое количество пациентов за короткий период времени — не такая уж и простая задача.
С другой стороны, небольшая выборка означает, что количество побочных эффектов препарата может быть значительно выше, чем известно на данный момент. Так, производитель уведомляет, что один из СМА-пациентов, который участвовал в клинических исследованиях за пределами США, через 12 дней после инфузии препарата начал страдать от дыхательной недостаточности. Также у него были зафиксированы лейкоэнцефалопатия, приступы гипотензии и судорог примерно через месяц после начала лечения. Через 52 дня наступил летальный исход. Но пока сложно сказать, является ли подобное развитие событий реакцией на введение препарата или же эти симптомы появились бы у СМА-пациента и без использования «Золгенсмы».
Важно также отметить, что долгосрочное влияние препарата на организм человека пока неизвестно. Прежде всего, не ясно, будет ли экспрессия гена SMN1 в организме пациента поддерживаться постоянно или постепенно сойдет на нет. Чтобы ответить на этот вопрос, «Новартис» обязана постоянно собирать данные долговременного наблюдения. В разрезе стоимости это является ключевым риском для плательщиков. Компания-производитель пытается снизить эти риски, предлагая рассрочку на пять лет пациентам, с правом приостановить выплаты, если препарат не будет проявлять своего терапевтического действия. Однако если экспрессия гена прекратится после истечения пятилетнего срока, то никакой финансовой компенсации за это не предусмотрено.
Не проводились исследования на животных по оценке онкогенного и мутагенного действий препарата, хотя есть данные о подобных исследованиях для вектора AAV9. Фактически, «Золгенсма» может оказаться как настоящей панацеей для людей, больных СМА, так и генетической бомбой замедленного действия, эффект которой будет заметен только через годы или десятилетия.
Следующий шаг
Компания-производитель «Золгенсмы» планирует в будущем применять препарат для пациентов разных возрастов со СМА II и III типов. Также «Новартис» работает над регистрацией препарата за пределами США.

Рисунок 5. Рисдиплам — препарат для лечения спинально-мышечной атрофии, который находится на стадии клинических испытаний на людях
Появление «Золгенсмы» на фармакологическом рынке повлияло на продажи «Спинразы», поэтому компания «Биоген» уже проводит клинические исследования, направленные на усиление терапевтической активности «Спинразы» путем увеличения ее дозы [11].
Но «Спинраза» и «Золгенсма», возможно, недолго будут единственными препаратами для лечения спинально-мышечной атрофии. Компания «Рош» (Roche) уже достаточно давно ведет клинические испытания своего лекарственного средства (рисдиплама) и, по всей видимости, в ближайшие пару лет данный препарат также выйдет на фармацевтический рынок (рис. 5).
Рисдиплам, как и нусинерсен («Спинраза»), не влияет на ген SMN1, а модифицирует сплайсинг мРНК гена SMN2. Однако у рисдиплама есть существенное отличие: препарат принимается перорально и не требует введения в спинно-мозговую жидкость. Кроме того, рисдиплам подходит для всех типов СМА и в клинических испытаниях показывает более высокую эффективность, чем нусинерсен [12].
По прогнозам экспертов, цена препарата будет значительно ниже, чем «Спинразы» и «Золгенсмы», поэтому новое средство сможет составить серьезную конкуренцию уже существующим лекарствам [8].
Уже в конце 2019 года компания Roche планирует подать документы на одобрение препарата в FDA и EMA (Европейское медицинское агентство) [13].
Сегодня «Золгенсма» является одним из немногих одобренных генотерапевтических препаратов, и единственный — для лечения СМА. Данная технология является чрезвычайно перспективной и теоретически может подарить шанс СМА-пациентам на продолжительную жизнь высокого качества. Однако возможные побочные эффекты и высокая стоимость препарата пока не позволяют делать поспешных радужных выводов.
Источник


