Технологии создания лекарственных средств
Разработка лекарственного средства начинается с синтеза новых химических соединений. Вещества со сложной структурой можно получить из различных иа очников, например растений (сердечные гликозиды), тканей животных (гепарин), микробных культур (бензилпенициллин), культур человеческих клеток (урокиназа), или посредством генно-инженерных технологий (человеческий инсулин).
Чем больше ясности во взаимоотношениях структуры и активности, тем более направленным оказывается поиск новых веществ.
а) Доклинические испытания дают информацию о биологических эффектах новых веществ. Начальный скрининг может включать биохимические и фармакологические исследования (анализ связывания с рецептором) или эксперименты на культурах клеток, изолированных клетках и изолированных органах.
Поскольку эти модели не способны воспроизвести сложные биологические процессы, происходящие в интактных организмах, любое потенциальное лекарственное средство необходимо проверить на животных. Только эксперименты на животных позволяют выяснить, возникаютли желаемые эффекты при дозах, не вызывающих токсичности или сопровождающихся слабой токсичностью. Цель токсикологических исследований заключается в том, чтобы оценить:
1) токсичность, обусловленную кратковременным или длительным приемом;
2) генетические повреждения (генотоксичность, мутагенез);
3) развитие опухолей (канцерогенность);
4) возникновение врожденных дефектов (тератогенность).
В экспериментах на животных также оценивают всасывание, распределение, метаболизм и элиминацию (фармакокинетика) изучаемых веществ. На уровне доклинического изучение лишь у малой части новых веществ обнаруживается потенциал для применения у человека.
Фармацевтическиетехнологии предлагают методы изготовления лекарственных форм.
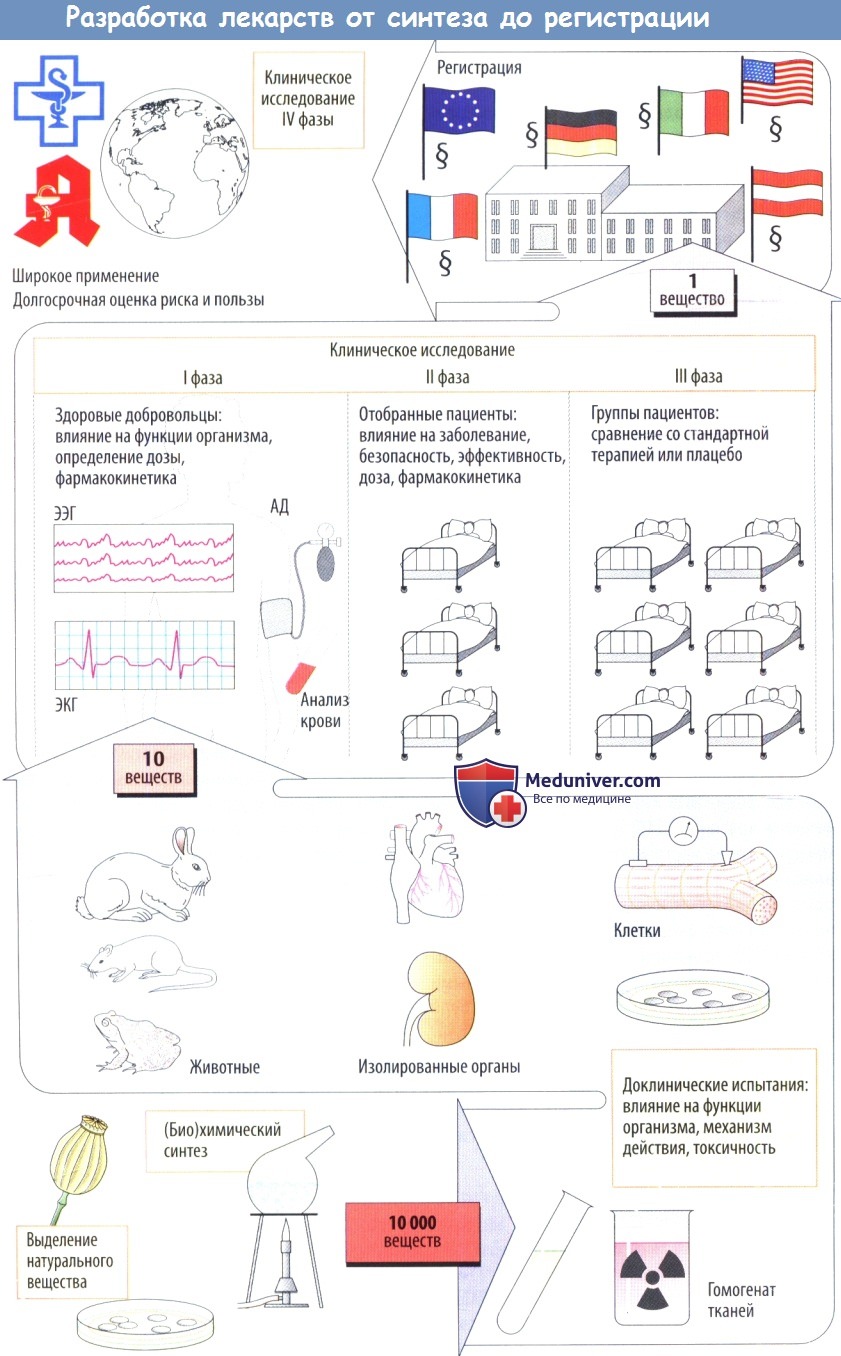
б) Клинические испытания начинаются с исследований I фазы, в которых участвуют здоровые лица; цель этих исследований — определить, будут ли эффекты, наблюдаемые у животных, также возникать у людей. Кроме того, на данном этапе определяется дозозависимость клинических эффектов.
Во II фазе потенциальные лекарственные средства сначала проверяют на отобранных пациентах на терапевтическую эффективность при заболевании, для лечения которого эти препараты предназначались. Если полезное действие очевидно, а частота побочных эффектов приемлема, начинается III фаза, в которой участвует более крупная группа пациентов, у которых новое средство сравнивают с традиционными методами лечения сточки зрения терапевтического результата.
Как форма экспериментов на людях, эти клинические исследования подвергаются анализу и одобрению этическими комитетами медицинских учреждений в соответствии с международными правилами проведения (Хельсинкской, Токийской и Венецианской декларациями). Во время клинических исследований выясняется, что многие вещества нельзя использовать. Как правило, в конце концов примерно из 10 000 вновь синтезированных веществ остается только одно.
в) Решение зарегистрировать новое лекарственное средство выносится национальным регуляторным органом (Food and Drug Administration в США, Health Protection Branch Drugs Directorate в Канаде, комиссией ЕС вместе с European Medicines Agency в Великобритании), которому производители должны подавать регистрационные документы.
Заявители должны документально подтвердить результатами соответствующих испытаний (доклинических и клинических), что критерии эффективности и безопасности удовлетворены и что лекарственные формы продукта (таблетки, капсулы и т. д.) соответствуют всем стандартам контроля качества.
После регистрации новое лекарственное средство может продаваться под торговым названием, оно должно быть доступным, выписываться врачами и отпускаться фармацевтами. В это время наблюдение продолжается в форме постмаркетинговых исследований (IV фаза клинических исследований)
г) Фармакологический надзор — действия, направленные на то, чтобы выявлять и устранять связанные с препаратом риски во время проведения клинических исследований и последующего его выхода на рынок. Фармаконадзор включает отчеты о предполагаемых случаях нежелательных реакций, направляемые в национальные регуляторные органы.
На основе длительного опыта применения можно правильно оценить соотношение риска и пользы и, следовательно, терапевтическую ценность нового лекарственного средства. Если новый препарат имеет небольшое преимущество перед существующими, необходимо принимать во внимание соотношение затрат и пользы от применения лекарственного средства.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Источник
Как создать препарат в лаборатории и перенести его в производство
Все мы ходим в аптеку и покупаем лекарства: таблетки, порошки, мази, растворы и многие другие формы лекарственных веществ. Но мало кто знает, как происходит создание лекарств и какой путь необходимо пройти от научной разработки в руках ученого до получения регистрационного досье на готовый препарат.
Разработка и создание лекарственных препаратов проходят при финансовой поддержке различных государственных и коммерческих структур (фондов) в соответствии с утвержденными приоритетными направлениями развития науки, технологий и техники в Российской Федерации, согласно перечню (указ президента России от 7 июля 2011 года №899). Одним из таких направлений являются «Технологии снижения потерь от социально значимых заболеваний».
Терапия и диагностика онкологических заболеваний — одно из приоритетных направлений. Многие ученые работают над созданием новых низкомолекулярных веществ для химиотерапии, получением новых аналогов уже существующих препаратов для преодоления возникающей резистентности опухолевых клеток, а также созданием новых лекарственных форм препаратов для улучшения биодоступности активного компонента и уменьшения побочных эффектов. В последние годы широко развивается направление адресной доставки препаратов — например, на основе антител.
Нашей научной группой под руководством члена-корреспондента РАН, профессора, доктора химических наук Евгения Северина разработан универсальный подход к созданию препарата для адресной доставки в злокачественные новообразования. Данный метод заключается в синтезе наночастиц, содержащих активное вещество, с последующей конъюгацией векторной молекулой — белка, способного связываться с рецепторами на поверхности опухолевых клеток. Предварительные исследования показали многообещающие результаты, проведенные доклинические испытания подтвердили, что разработанный прототип препарата обладает большей противоопухолевой активностью по сравнению с аналогом, представленным на рынке. Суммируя опыт проведения таких исследований, мы можем описать стандартный протокол проведения исследований при разработке нового препарата и оформления нормативной документации.
Каков же общий путь исследований, позволяющих провести доказательную базу эффективности и безопасности нового препарата, и какое количество времени для этого необходимо? После проведения этапов разработки подхода создания препарата и проведения предварительных экспериментов для доказательства его эффективности коллективом оформляется заявка для участия в конкурсе для предоставления финансирования на проведение доклинических испытаний. Стандартный грант предоставляется на три года, по результатам выполнения которого у группы исследователей, выполняющих данную работу, будет готовый прототип препарата, изучена его эффективность, безопасность и оформлены все необходимые нормативные документы, на основе которых формируется досье и подается на рассмотрение в Министерство здравоохранения России.
Первое, что необходимо сделать при разработке нового препарата,— проведение обширного литературного и патентного поиска в близких и смежных областях, чтобы избежать «изобретения велосипеда». Если патентная чистота подтверждена, можно приступать к экспериментальной работе.
В полученном гранте на проведение доклинических испытаний прописан календарный план и список этапов, которые необходимо выполнить, а затем подготовить отчетную документацию. Все этапы регламентированы соответствующими нормативными документами. Настольными книгами для специалистов являются «Государственная фармакопея Российской Федерации» и «Руководство по проведению доклинических исследований лекарственных средств» под редакцией А. Н. Миронова. В фармакопее прописаны все требования и нормы к разрабатываемым препаратам, какие виды исследования необходимо провести для подтверждения состава, структуры и свойств будущего лекарства или новой лекарственной формы (порошки, таблетки, растворы и пр.). В руководстве по проведению доклинических исследований подробно изложено, как необходимо проводить доклинические испытания, чтобы исследование было стандартизировано: выбор вида животных, их количества, кратность введения, дозы и пр.
Для проведения такого широкого спектра исследований необходимо соответствующее количество будущего препарата. В лабораторных условиях обычно отрабатывают технологические режимы и оптимальные параметры получения — от температурного режима до масштабирования процесса — и изучают влияние этих параметров на свойства получаемого продукта. По оптимизированным условиям пишут лабораторный регламент, где четко описано, как именно и при каких условиях необходимо проводить каждый этап получения препарата и что должно быть на выходе, вплоть до учета потерь производства. Лабораторный регламент является официальным нормативным документом, на его основе составляют опытно-промышленный и промышленный регламенты. Для последних двух необходима специальная производственная площадка, аттестованная под изготовление похожего продукта, как и разработанный препарат (рекомбинантные белки, вакцинные препараты, противоопухолевые препараты и т. д.). Необходимо перенести лабораторную технологию получения продукта в больший масштаб на промышленное оборудование, отработать регламентированный процесс получения и оптимизировать технологию с учетом новых объемов. Таким образом, на промышленных производственных площадках по опытно-промышленному и промышленному регламенту получают опытные партии препарата, которые затем необходимо проверять на соответствие всем показателям, которые были установлены разработчиками после получения оптимальной партии по лабораторному регламенту. Все требования к полученному препарату описаны в нормативном документе — фармацевтической статье предприятия (ФСП).
ФСП пишут по тем методам, с помощью которых анализируют полученный препарат в соответствии с государственной фармакопеей. Для включения метода в ФСП необходимо использовать либо стандартизированные методы, приведенные в фармакопее, либо валидированные методы, которые были использованы, но отсутствуют в фармакопее или отличаются по условиям проведения от описанных. Чем больше активных (целевых) и вспомогательных компонентов в препарате, тем больше методов содержит ФСП. Необходимо провести количественный анализ активного компонента и примесей, полный качественный анализ, а также подтвердить сохранение функциональной активности основного компонента.
Зачем нужна фармацевтическая статья предприятия? В технологическом процессе получения препарата могут возникнуть непредвиденные неполадки на какой-либо стадии производства. Для выявления несоответствия продукта (брака производства) необходимо проводить анализ каждой партии. Если была получена бракованная партия, ее можно будет легко выявить, проведя все анализы и сравнив с установленными нормами в ФСП.
После получения опытных партий необходимо изучить стабильность полученного препарата, чтобы доказать, что за указанный промежуток времени (срок годности) не происходит никаких существенных изменений и свойства будущего лекарства остаются неизменными. Стандартный срок хранения лекарственных средств — от полугода до трех лет. Для противоопухолевых препаратов — два года. Для конкурентоспособности и рентабельности будущего лекарства необходимо придерживаться таких же сроков годности, а также ориентироваться на аналоги. Однако ждать два года, чтобы узнать, стабилен ли препарат и проходит ли он по всем нормам и стандартам, нет необходимости. Существуют протоколы, описанные в фармакопее (ОФС.1.1.0009.15 «Сроки годности лекарственных средств»), позволяющие сократить период исследования до года и даже шести месяцев, используя более агрессивные условия исследования. Если препарат по всем показателям сохраняет количественные, качественные и функциональные характеристики, указанные в ФСП, следующий этап — проведение доклинических исследований на животных. На все перечисленные стадии оформляют нормативные документы: регламенты на производство, отчеты о валидации, акты наработки, ФСП, протоколы анализа партий, подтверждающие соответствие полученного продукта описанному в документах.
Для изучения безопасности и эффективности полученного препарата на лабораторных животных утверждается план доклинических исследований, в котором перечислены все этапы исследования и их последовательность. План и модели проведения исследований составляются в зависимости от типа разработанного препарата и описаны в «Руководстве по проведению доклинических исследований лекарственных средств». Для оформления регистрационного досье необходимо провести полные доклинические исследования: исследование нескольких видов токсичности (общетоксическое действие, аллергизирующие свойства, иммунотоксическое действие, репродуктивная токсичность и др.), эффективности действия (например, противоопухолевый эффект нового препарата в сравнении с аналогами), изучить фармакокинетику. Все полученные данные обрабатывают статистически и оформляют в нормативный документ — отчет о доклинических исследованиях с прикреплением первичных результатов. На основании полученных данных, в случае если препарат обладает эффективностью и при этом безопасен для применения, разработчики пишут план проведения первой стадии клинических испытаний, проект инструкции по применению и проект брошюры исследователя. Из составленных документов формируется регистрационное досье, которое и подается на рассмотрение в Министерство здравоохранения России с другими сопутствующими документами.
До последнего этапа, описанного в данной статье, доходят немногие разработки. Путь от научной идеи до регистрации может занимать от трех лет до десятилетий. При наличии оформившейся идеи, прошедшей предварительные фундаментальные исследования, все описанные этапы исследования можно провести за три-пять лет. А дальше препарат ждет еще более сложный, но не менее интересный путь: клинические испытания, регистрация, производство и выход на рынок — при условии наличия хороших результатов на этапе клинических испытаний.
Елена Никольская, кандидат химических наук, старший научный сотрудник ИБХФ РАН
Источник
Долгий путь от молекулы до лекарства: как пройти его максимально эффективно
Поиск инноваций – неотъемлемая часть фармацевтического бизнеса. Технологии в фармацевтике развиваются стремительно: в будущем лекарства можно будет выпускать для конкретного человека на основе его генетического паспорта. Но об их доступности говорить рано — в среднем на разработку одного лекарства тратится от 1 до 2,5 млрд долларов и около 10–15 лет. Путь от молекулы в лабораторной пробирке до лекарственного препарата невероятно тернист и долог. И он может прерваться на любом этапе: из всех веществ, участвующих в доклинических разработках, лишь 2% становятся зарегистрированными препаратами. Остальные оказываются недостаточно эффективными или вызывают слишком тяжелые побочные эффекты. О том, как создаются лекарства и команды разработчиков, а также о новых подходах к R&D в фармацевтике рассказывает к.м.н., медицинский директор АО «Байер» и руководитель Медицинского кластера стран СНГ Дмитрий Власов.
Фото предоставлено пресс-службой компании Bayer
В классическом понимании лекарственный препарат — это химическое соединение с определенными свойствами. Сейчас это понятие расширилось, поскольку кроме малых молекул, полученных химическим синтезом, фармацевты используют и большие активные молекулы белков – например, антитела.
От биомишени – до мышей
В создании лекарства все начинается даже не с молекулы, а с определения биологической мишени. Это некий белок, который играет важную роль в том или ином заболевании. Чтобы бороться с симптомами болезни необходимо воздействовать на этот белок – либо повышая, либо снижая его активность. На эту мишень и должно быть направлено создаваемое лекарство.
Следующий этап – выбор соединения, которое будет действовать на данный белок.
Сейчас самый ранний этап разработки проходит с применением компьютерных технологий. Это значительно дешевле и проще – отскринировать большое количество молекул виртуально. С помощью компьютерного моделирования оценивается взаимодействие биомишени и целого ряда химических соединений. Оценке подвергается способность вещества соединяться с биомишенью в нужном месте и с необходимой силой.
Третий этап – настоящий «живой» скрининг.
После отбора наиболее эффективных молекул их действие тестируется на живых объектах, для начала – на клетках. К примеру, для разработки противоопухолевых препаратов тестируемые соединения испытываются на специализированных клеточных линиях, представляющих собой клетки опухолей различного типа. В этом случае оценивается уже непосредственно эффект воздействия соединения на клетку, например, его способность остановить или замедлить неконтролируемое деление раковых клеток. Как и в первом этапе, по результатам экспериментов отбираются наиболее перспективные соединения-лидеры.
Эти соединения уже доказали свою эффективность, однако и их можно и нужно дополнительно улучшить с помощью химических модификаций. На примере каждого перспективного соединения создают целый пул молекул, похожих по структуре, но имеющих небольшие отличия, которые могут влиять положительно на такие параметры молекулы, как эффективность (сильное воздействие на биомишень), безопасность (снижение токсичности), фармакокинетику (время жизни в организме). Полученный пул молекул снова тестируют на клеточных моделях. Этот процесс называется оптимизацией лидерных соединений.
Следующий этап – доклинические исследования, проверка действия препарата на уровне целого организма. Стандартным объектом экспериментов на данном этапе являются лабораторные животные, чаще всего – мыши или крысы. На животных воссоздается модель заболевания (например, стимулируется появление опухоли) и тестируются будущие лекарственные препараты. На этапе доклинических исследований необходимо понять не только эффективность действия вещества на уровне организма (которая может значительно отличаться от таковой на уровне тестирования на клетках), но и изучить такие важнейшие параметры как фармакокинетику, токсичность (опасность для жизни и здоровья организма), оценить динамику распределения и накопления вещества в органах и тканях.
Крайне важно учитывать, что на следующем этапе в исследованиях будут участвовать уже люди, поэтому необходимо исключить максимум рисков с точки зрения безопасности (для нивелирования или минимизации вреда для человека), так и эффективности (исследования на животных значительно дешевле, чем исследования на человеке. Так что, гораздо проще отсеять неэффективные препараты на данном этапе).
Качество данных на этапе доклинических исследований имеет огромное значение, поэтому для всех центров доклинических исследований существуют правила надлежащей лабораторной практики (Good Laboratory Practice или GLP), которые содержат стандарты к содержанию животных и проведению доклинических исследований. Оценку соответствия центров доклинических исследований стандартам GLP проводят независимые сертифицирующие организации.
Когда получены все данные по эффективности, безопасности и фармакокинетике и отобраны лучшие соединения, можно переходить к следующему, самому ответственному этапу.
Клинические исследования
Основные затраты времени и ресурсов занимают доклинические и клинические исследования, в том числе – проверка токсичности, подбор оптимальной дозировки и режима лечения, а также отработка промышленной технологии производства, выбор оптимальной лекарственной формы.
Если проведение доклинических исследований – зона ответственности научных лабораторий институтов или фармацевтических компаний (хотя сами лаборатории, как мы помним, сертифицируются по стандартам GLP), то на этапе клинических исследований значительно возрастает роль государства. Прежде чем начать клиническое исследование, необходимо получить разрешение регулирующего органа, которое он выдает на основании анализа данных о препарате, представляемых фармацевтической компанией.
Все клинические исследования состоят из трех фаз. На первой фазе идет оценка безопасности препарата. Эти исследования часто проводят на здоровых добровольцах, за исключением случаев, когда препарат заведомо высокотоксичен (как, например, почти все противоопухолевые препараты). В этом случае препарат при условии согласия могут уже принимать больные пациенты, которым не помогли другие методы лечения. Для них участие в исследованиях – серьезный шанс продлить жизнь и, возможно, заметно улучшить состояние здоровья. Кроме безопасности препарата, на этом этапе также оценивается фармакокинетика вещества в организме человека.
Вторая фаза – оценка эффективности и выбор оптимальной дозировки препарата. На этом этапе препараты всегда исследуются на пациентах с определенным заболеванием. Помимо оценки эффективности, безусловно, оценивается и безопасность (препарат может по-разному действовать на здорового и больного человека).
Третья фаза – масштабное исследование препарата с учетом полученных ранее результатов. Нужно подтвердить, что лекарственное средство дает не кратковременный эффект, а достоверно улучшает важные для жизни параметры. Очень важным является вопрос всесторонней проверки безопасности лекарственных веществ, изучение побочных явлений их применения. «Современные лекарства подобны ядерной энергии: они могут приносить огромную пользу и огромный вред», — в свое время сказал Деррик Данлоп, первый председатель комиссии по безопасности лекарств Великобритании. Сегодня эти слова не утратили своей актуальности.
Третья фаза – самая длительная и дорогостоящая. Она может стоить сотни млн долларов и охватывать несколько десятков тысяч пациентов в разных странах. По итогам третьей фазы препарат получает регистрацию и может быть использован для лечения на коммерческой основе.
Далее возможна четвертая фаза – исследования на предмет расширения действия препарата в отношении других заболеваний, или получение дополнительных сведений о безопасности препарата. Здесь также ведется мониторинг долгосрочной результативности.
Предусмотреть последствия
Часто бывает, что тестируемое лекарство эффективно для животных, но на организм человека не действует. Экспертам важно предусмотреть такой вариант заранее – до клинических исследований. Для этого используют подход под названием трансляционная медицина. На подопытном животном разрабатывают релевантную модель болезни типичную для организма человека и исследуют будущее лекарство.
В этом случае данные об эффективности с большей вероятностью совпадут с результатами клинических исследований. Если, конечно, исследователи проведут все расчеты точно. Здесь многое зависит и от уровня компетенции экспертов, команды ученых. Поэтому тщательное отношение к квалификации сотрудников, постоянное ее повышение и правильная система развития и мотивации – одна из важнейших задач фармацевтической компании.
Открытые инновации
Если посмотреть на статистику расходов фармкомпаний, то можно увидеть, что на R&D они тратят с каждым годом все больше (по оценкам Evaluate Pharma в ближайшие 5 лет общие затраты фармкомпаний на R&D будут расти в среднем на 3% в год и к 2022 году достигнут более $180 млрд). В научные разработки уходят миллиарды долларов. При этом количество зарегистрированных молекул в мире остается примерно одинаковым. Получается, стоимость каждой новой молекулы возрастает. И, судя по всему, исследования и разработки новых препаратов в ближайшие годы дешевле не станут.
Понятно, чтобы закрыть все вопросы в цепочке создания лекарств с учетом нынешних объемов расходов ни у одной компании не хватит ни человеческих, ни финансовых ресурсов.
На помощь здесь приходит концепция открытых инноваций – вовлечение внешних сторон в процесс исследований и разработок. На пути создания новых медицинских решений фармкомпании активно взаимодействуют с научным сообществом, стартапами и институтами развития. Выделяют гранты на поддержку разработок в том или ином направлении. Либо делятся с институтами своей экспертизой, лабораторной коллекцией химических соединений и антител.
Возникает ситуация сотрудничества, когда компания помогает ученым и исследователям повысить вероятность успешности их разработок, а они помогают фармацевтике за счет новых идей, подходов, решений. Такое сотрудничество неизбежно: если бы компании вели все разработки самостоятельно в своих лабораториях, то затраты были бы в разы больше.
За счет оптимального расходования ресурсов модель открытых инноваций позволяет исследовать максимальное количество технологий.
В то же время эта модель приносит дополнительный доход и новые возможности университетам, где была создана исходная молекула. Даже если проекты, с которыми мы работаем, окажутся неуспешными, будет сформирована экосистема, коллективы, которые знают, как надо действовать дальше. Если у них не получится с этим проектом, то будет успех со следующим. В глобальном смысле это воспитание некой культуры разработок инновационных препаратов, что очень важно для развития медицины.
На Западе эта модель давно и успешно развивается. Университеты и научные институты ведут множество исследований для фармкомпаний.
Но в России модель открытых инноваций пока находится в процессе становления. Да, отечественные компании объявляют конкурсы по поиску новых технологий, но не обкатан метод взаимодействия с разработчиками на дальнейшем этапе. Бывают, ученые демонстрируют интересные перспективные разработки. Казалось бы, вот он – прорыв в медицине! Нужно лишь довести идею до конца. Но, к сожалению, со многими проектами так и не выстраиваются долгосрочные истории взаимодействия.
Как развить научные фарм-стартапы в России
Как показывает практика, многие фундаментальные идеи в фармацевтике при определенной поддержке можно перевести в сферу реального бизнеса. И речь здесь идет не только об инвестициях. Лет десять назад у российских разработчиков было две проблемы: где найти финансирования и как эффективно использовать полученные средства. С первой проблемой при помощи господдержки, профильных грантов, венчурного инвестирования понемногу научились справляться. Но уверенное понимание того, как при наличии разработок заниматься не только развитием науки, но и созданием коммерчески успешного проекта, еще предстоит сформировать.
И здесь очень важен опыт тех, кто уже прошел путь вывода препарата или медуслуги на рынок – более крупных фармкомпаний, экспертов в области бизнеса. В США или Европе, например, при ведущих университетах есть профильно-работающие центры с представителями потенциальных инвесторов, которые позволяют разработчикам структурировать проекты и должным образом их направлять. Распространена и практика бизнес-инкубаторов, которые содействуют молодым ученым в создании собственных медико-биологических стартапов. Могу рассказать о примере Bayer. С 2012 года мы запустили собственную модель лаборатории-инкубатора для фармстартапов – КоЛаборатор. Поначалу в США и Европе, а сейчас и в России. Суть программы: команды проходят конкурсный отбор и, попадая в инкубатор, получают доступ к обширной лабораторно-офисной инфраструктуре, профессиональным знаниям и глобальной исследовательской сети фармкорпорации. В свою очередь, мы рассчитываем стать первыми к кому новые компании с уникальными разработками обратятся в поиске возможных партнеров. При этом, сами стартапы остаются независимыми.
В России участниками такого бизнес-инкубатора могут стать проекты образованные студентами, аспирантами, преподавателями и научными сотрудниками ведущих университетов страны, а также высокотехнологические стартапы, занимающиеся разработкой в области фармацевтики.
Конечно, у нас модель КоЛаборатора имеет некоторые отличия. В России мы в большей степени предоставляем экспертную поддержку стартапам, как в научной, так и в коммерческой сфере – то, чего им больше чего не хватает. Планируем расширять партнерства с ключевыми центрами в Москве, Санкт-Петербурге, в Новосибирске и других городах с большим научным потенциалом.
На данный момент резидентами КоЛаборатора являются три стартапа. В рамках КоЛаборатора Bayer предоставляет международную экспертизу в области исследований и разработок, а также делится технологиями развития инновационных решений.
Среди участников КоЛаборатора проект в области терапии хронической тромбоэмболической легочной гипертензии. Это разработка научного коллектива Национального медицинского исследовательского центра имени В. А. Алмазова – партнера проекта КоЛаборатор.
Второй проект – ученых Новосибирского института молекулярной и клеточной биологии. Они разрабатывают метод клеточной терапии в онкологии, который позволит в перспективе эффективно бороться с такими распространенными заболеваниями как рак простаты, рак молочной железы и рак легких.
Третий проект также касается сердечно-сосудистой системы. Это разработка компании Target Medicals, резидента «Сколково», воздействующая на гормональную систему регуляции кровяного давления (РААС-систему).
Ведущие эксперты Bayer консультируют участников КоЛаборатора на всех этапах деятельности, включая исследования, разработки, позиционирование будущего продукта на рынке. По каждому проекту мы выстраиваем отдельную стратегию поддержки, развития бизнеса. В этом году мы согласовали выделение небольших грантов нашим резидентам. Но основная цель для участвующих проектов – долгосрочная поддержка проектов с помощью экспертизы компании. Проекты должны получать ее на всех этапах развития, начиная с экспертизы в области медицинской химии, и заканчивая стратегией выхода препарата на рынок, стратегией патентования, возможного конкурентного анализа, рыночного позиционирования.
Задача – сформировать эффективную цепочку создания лекарств и коммерциализации разработок в России – непростая. Но перспективы и возможности для этого есть. Судя по вниманию глобального фармбизнеса к R&D в России, а также задачам, обозначенным Президентом России, в рамках стратегии развития научно-исследовательского потенциала нашей страны, такое утверждение представляется абсолютно реалистичным.
Источник


