- Документы, предоставляемые в Росздравнадзор для подтверждения качества лекарственного препарата, фармацевтической субстанции, биологического материала или иного вещества в составе МИ
- История вопроса
- Какие же документы нужны?
- Документ производителя, подтверждающий качество лекарственного средства промышленной серии или партии (протокол анализа или сертификат анализа)
- Документация по качеству с описанием методов контроля
- Копия лицензии на производство лекарственного средства, выданной уполномоченным органом страны-производителя
- Копия заключения о соответствии производителя лекарственного средства требованиям правил надлежащей производственной практики, выданного уполномоченным органом страны-производителя (при наличии)
- Дата государственной регистрации лекарственного препарата и его регистрационный номер в государственном реестре лекарственных средств для медицинского применения (при наличии)
- Сертификат GMP: подтверждение качества лекарственных средств
- К каким производствам применима эта процедура?
- Нормативная база
- Преимущества обладания сертификатом
- Стандарт GMP в международной практике
- Правила GMP в России
- Процедура получения сертификата в России
- Документы для сертификации
- Сроки сертификации
- Стоимость получения сертификата
Документы, предоставляемые в Росздравнадзор для подтверждения качества лекарственного препарата, фармацевтической субстанции, биологического материала или иного вещества в составе МИ

К нам часто обращаются с вопросом о том, какие документы дополнительно необходимо предоставлять в Росздравнадзор для выполнения требований подпункта «н» пункта 10 Правил государственной регистрации медицинских изделий (Постановление Правительства РФ №1416).
История вопроса
До июня 2018 года медицинские изделия, содержащие лекарственные средства (фармацевтические субстанции или лекарственные препараты) в принципе невозможно было зарегистрировать, если лекарственное средство не было зарегистрировано в России. Это создавало сильные трудности для многих участников сферы обращения медицинских изделий, в особенности в областях косметологии и стоматологии.
В марте 2018 года ООО «МЕДРЕЛИС» была подготовлена инициатива с предложением внесения изменений в действующие нормативно-правовые акты. Инициатива была поддержана многими участниками сферы обращения медицинских изделий, и по ее результатам состоялась встреча инициативной группы с представителями Росздравнадзора и Министерства здравоохранения РФ.
В итоге 31 мая 2018 года было принято Постановление Правительства РФ №633, вносящее правки в Правила государственной регистрации медицинских изделий. Так, в том числе, перечень документов, предоставляемых в Росздравнадзор при регистрации дополнился подпунктом «н» следующего содержания:
«н) копии документов, подтверждающих качество лекарственного препарата, фармацевтической субстанции, биологического материала и иного вещества, с использованием которых произведено медицинское изделие или которые входят в его состав и которые предназначены для применения только с учетом назначения медицинского изделия, определенного производителем, и выданных в соответствии с законодательством страны происхождения лекарственного препарата, фармацевтической субстанции, биологического материала и иного вещества»
Таким образом, в составе медицинских изделий стало возможно использовать не зарегистрированные в РФ лекарственные средства, при условии предоставления соответствующих подтверждающих документов. Недавно Росздравнадзор конкретизировал эти документы, но эта конкретизация все равно не внесла достаточную ясность. Мы хотели бы взять на себя смелость прокомментировать ответ Росздравнадзора, показав примеры из нашего успешного опыта регистрации таких медицинских изделий.
Какие же документы нужны?
Итак, Росздравнадзор указывает, что к таким документам могут относиться:
Документ производителя, подтверждающий качество лекарственного средства промышленной серии или партии (протокол анализа или сертификат анализа)
Сертификат анализа (Certificate of Analysis, COA) – это документ, который производитель составляет по результатам выходного контроля конкретной партии лекарственного средства (или иного вещества). Большинство лекарственных средств зарубежного производства имеет такой документ. В различных странах он регламентируется различными документами, но основная информация, которую такой документ содержит, остается неизменной. Мы хотели бы разобрать содержание сертификата анализа на примере законодательства Европейского Союза, где оно регламентируется разделом 11.4 документа EU GMP Guide Part II. Согласно требованиям этого документа, сертификат анализа содержит:
- Name of the intermediate or API (наименование вещества)
- Batch number (номер партии)
- Release date (дата выпуска)
- Expiry date (дата истечения срока годности)
- List of the tests performed including acceptance limits (перечень проведенных испытаний, включая критерии приемлемости)
- Numerical results (количественные результаты)
- Dated signature by authorised personnel (дата и подпись уполномоченного лица)
- Name of the manufacturer (наименование производителя)
- and Name of the laboratory (и наименование лаборатории)
Пример сертификата анализа можно просмотреть ниже:
 Источник: http://www.humate.info/fertilizer-edta-zn-15.html
Источник: http://www.humate.info/fertilizer-edta-zn-15.html
Документация по качеству с описанием методов контроля
В качестве такой документации мы видим паспорт безопасности (Material Safety Data Sheet, MSDS, SDS). Он составляется, как правило, на продукцию, которую с точки зрения зарубежного законодательства можно отнести к «иным веществам» (например, хлорид натрия, бриллиантовый синий, оксид кремния). Важно отметить, что паспорт безопасности выпускается не на конкретную партию вещества, а на вещество в целом.
Рассмотрим, опять же, пример Европейского Союза. В нем содержание паспорта безопасности регламентировано следующими документами:
1. Регламент (ЕС) №1907/2006 от 18.12.2006 г. Европарламента и Совета Европы, касающийся правил регистрации, оценки, санкционирования и ограничения химических веществ (REACH);
2. Регламент (ЕС) №453/2010 Европарламента и Совета Европы о внесении поправок в Регламент (ЕС) 1907/2006 от 18.12.2006 г. Европарламента и Совета Европы, касающийся правил регистрации, оценки, санкционирования и ограничения химических веществ;
Кроме того, в Российской Федерации действует межгосударственный стандарт ГОСТ 30333-2007 «Паспорт безопасности химической продукции. Общие требования».
Требования к паспорту безопасности во всех вышеперечисленных документах идентичны. Так, он должен содержать следующие 16 разделов (более подробно с содержанием этих разделов можно ознакомиться в вышеуказанных документах):
1) Идентификация химической продукции и сведения о производителе или поставщике;
2) Идентификация опасности (опасностей);
3) Состав (информация о компонентах);
4) Меры первой помощи;
5) Меры и средства обеспечения пожаровзрывобезопасности;
6) Меры по предотвращению и ликвидации аварийных и чрезвычайных ситуаций и их последствий;
7) Правила хранения химической продукции и обращения с ней при погрузочно-разгрузочных работах;
8) Средства контроля за опасным воздействием и средства индивидуальной защиты;
9) Физико-химические свойства;
10) Стабильность и реакционная способность;
11) Информация о токсичности;
12) Информация о воздействии на окружающую среду;
13) Рекомендации по удалению отходов (остатков);
14) Информация при перевозках (транспортировании);
15) Информация о национальном и международном законодательстве;
16) Дополнительная информация.
Пример паспорта безопасности можно посмотреть нажав на кнопку:
Копия лицензии на производство лекарственного средства, выданной уполномоченным органом страны-производителя
Может быть различной в различных странах. Выдается компетентным государственным органом (например, Food and Drug Administration (FDA) в США, Food and Drugs Control Administration (FDCA) в Индии и т.д.
Пример лицензии на производство лекарственного средства можно посмотреть ниже:

Копия заключения о соответствии производителя лекарственного средства требованиям правил надлежащей производственной практики, выданного уполномоченным органом страны-производителя (при наличии)
В качестве такого документа можно рассматривать сертификат GMP (Good Manufacturing Practice). Этот документ показывает, что система менеджмента качества производителя лекарственного средства соответствует Правилам GMP. Фактически, этот документ аналогичен сертификату соответствия требованиям стандарта ISO 13485 с тем отличием, что требования ISO 13485 применяются к медицинским изделиям, а Правила GMP – к лекарственным средствам.
Отметим, что бывают случаи, когда сертификат GMP получают и компании, производящие медицинские изделия, однако, такие случаи достаточно редки.
Пример сертификата GMP можно посмотреть ниже:

Дата государственной регистрации лекарственного препарата и его регистрационный номер в государственном реестре лекарственных средств для медицинского применения (при наличии)
Для лекарственных препаратов и фармацевтических субстанций, зарегистрированных в России. В этом случае достаточно просто указать эти данные в технической документации. Дополнительных документов предоставлять не надо.
Источник
Сертификат GMP: подтверждение качества лекарственных средств

Сертификат GMP — это соблюдение изготовителем лекарственных препаратов требований надлежащей производственной практики. В России они сформулированы в национальном стандарте ГОСТ Р 52249-2009, который идентичен правилам, действующим в Европейском Союзе.
К каким производствам применима эта процедура?
В настоящее время в странах, которые контролируют соответствие стандарту GMP на своих территориях, его правила применяются для проверки качества следующих категорий продукции:
- лекарственные препараты;
- медицинские изделия различного назначения, включая те из них, которые применяются в диагностических целях;
- продукты питания и ингредиенты для их производства;
- биологически активные добавки.
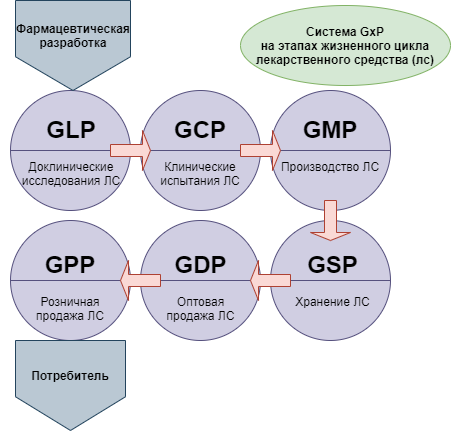
При этом для понимания ситуации следует принимать во внимание, что новая версия сертификации GMP — это не единственная система требований, которые в международной практике применяются в целях стандартизации медицинского обслуживания населения. Кроме них, производителям, работающим в такой сфере как фармация, необходимо соответствовать требованиям комплекса правил, объединенных под общим наименованием GxP:
- GLP — Good Laboratory Practice (надлежащая лабораторная практика);
- GCP — Good Clinical Practice (надлежащая клиническая практика);
- GDP — Good Distributon Practice (надлежащая дистрибьюторская практика);
- GACP — Good Agricultural and Collection Practice (надлежащая практика культивирования и сбора лекарственных растений).
Нормативная база
В Российской Федерации получение сертификата GMP осуществляется на основании действующей нормативной базы, включающей следующие основные правовые акты:
- национальный стандарт РФ ГОСТ Р 52249-2009, устанавливающий правила изготовления и контроля качества лекарственных препаратов;
- постановление Правительства от 5 июня 2008 года N 438 с рядом изменений, внесенных за последние годы, которое утверждает полномочия Министерства промышленности и торговли в этой области;
- постановление Правительства от 3 декабря 2015 года N 1314, устанавливающее порядок оценки соответствия производителей требованиям стандарта надлежащей практики;
- приказ Минпромторга от 14 июня 2013 года N 916, утверждающий правила применения надлежащей производственной практики в соответствии с актуальным стандартом;
- приказ Минпромторга от 26 мая 2016 года N 1714, определяющий административный регламент предоставления государственной услуги по выдаче документации, подтверждающей соответствие изготовителя установленным нормам надлежащей производственной практики;
- приказ Минпромторга России от 17.12.2015 N 4119, утверждающий правила ведения реестра сведений о том, какие лекарства имеют сертификат качества GMP в России.
При этом необходимо принимать во внимание, что в настоящий момент наша страна вместе с другими государствами, входящими в состав Евразийского экономического союза, находится на этапе становления общего рынка, объединяющего фармацевтическое и косметическое производство в границах Союза. Это предполагает в том числе введение в действие единых требований к качеству и безопасности таких продуктов. В соответствии с принятым в мире порядком они реализуются в форме внедрения стандартов надлежащей производственной практики. Применение таких стандартов регулируется следующими нормативными документами:
- Решение Совета ЕЭК от 3 ноября 2016 года N 77, утверждающее правила надлежащей производственной практики в границах ЕАЭС;
- Приказ Минпромторга от 4 сентября 2020 года N 2945, которым введен административный регламент предоставления госуслуги по выдаче документации, подтверждающей соответствие производств установленным правилам.
Обратите внимание!
Для полноценного применения разработанного административного регламента необходимо решение Правительства о порядке реализации некоторых процедур, связанных с проведением фармацевтических инспекций. Приказ № 2945 вступит в силу только после принятия соответствующего постановления: пока этого не произошло.
Преимущества обладания сертификатом
Несмотря на необходимость проведения достаточно сложной и дорогостоящей процедуры, производители знают, что сертификация по стандартам GMP является весьма важной для представителей фармацевтической отрасли. В частности, оно обеспечивает продукции и производству следующие серьезные преимущества:
- стабильное качество продукции, не зависящее от внешних факторов;
- повышение доверия потребителей, включая крупных оптовых покупателей, которые всегда отслеживают, какие производители имеют сертификат соответствия GMP на их продукцию;
- возможность вывода продукции на международные рынки, где ее может купить гораздо больше потребителей;
- возможность привлечения инвесторов для реализации проектов по расширению производства;
- получение преимуществ при участии в конкурсном отборе поставщиков, в том числе для государственных закупок.
КОММЕНТАРИЙ ЭКСПЕРТА АТТЭК
Каков срок действия сертификата?
Срок действия российских сертификатов составляет 3 года. При этом срок действия иностранного сертификата GMP составляет от 1 до 3 лет. По истечении этого периода сертификацию потребуется проходить заново. Кроме того, это означает, что на протяжении всего этого срока компании необходимо обеспечить соответствие своего производства и продукции требованиям комплекса правил GMP.
Кто в России занимается сертификацией по стандартам GMP?
Сейчас сертификация контролируется департаментом развития медицинской и фармацевтической промышленности Министерства промышленности и торговли РФ. Он является организацией, ответственной за обеспечение надлежащего контроля за качеством, безопасностью и эффективностью лекарственных средств. Осуществлением требуемых сертификационных процедур занимается Государственный институт лекарственных средств и надлежащих практик (ФГБУ «ГИЛС и НП»).
Стандарт GMP в международной практике
Процесс сертификации на соответствие лекарственного препарата стандартам GMP в международной практике имеет комплексный характер, а ее основной целью является подтверждение безопасности и действенности продукции. В этой связи для достижения поставленной цели специалисты аккредитованных сертификационных организаций не ограничиваются оценкой ряда выборочных образцов лекарственных препаратов, как это часто предусматривается другими стандартами. В процедуру установления требуемого уровня качества лекарств любой международный центр сертификации лекарственных средств включает оценку предприятия, занимающегося его выпуском. В результате эксперты, занимающиеся проведением сертификации, анализируют конкретный препарат и процесс его выпуска в следующих областях:
- оценка производства на соответствие критериям безопасности, включая проведение его проверки в отношении вероятности попадания в продукт посторонних примесей и веществ;
- оценка производства на соблюдение технических требований к выпуску продукции, включая выполнение условий относительно влажности, температуры и других параметров в производственных помещениях;
- оценка качества, безопасности и действенности лекарственных средств, производимых на конкретном предприятии;
- оценка соответствия параметров производства и характеристик лекарственного средства нормативной документации, принятой в рамках процедуры GMP.
Правила GMP в России
Порядок и сроки проведения всех операций в рамках этой процедуры, список лиц и организаций, ответственных за их осуществление, размер платы за проведение экспертной оценки и другие аспекты выполнения сертификации определены постановлением Правительства № 1314.
Процедура получения сертификата в России
Первым шагом для производителя, который желает пройти сертификацию, является подача соответствующего заявления в Минпромторг. В течение 10 рабочих дней специалисты ведомства проводят проверку корректности представленных в заявлении сведений и определяют возможность проведения сертификации.
В случае необходимости они вправе запросить у заявителя дополнительные документы, которые он обязан предоставить в течение 20 рабочих дней. В случае, если в отношении данного препарата принято положительное решение о проведении процедуры сертификации, необходимые данные направляются в ФГБУ «ГИЛС и НП», который в течение 20 рабочих дней с момента их получения обязан определить дату проведения сертификационных мероприятий и внести ее в график. Такая дата должна наступить не позднее 160 рабочих дней со дня, когда специалисты Минпромторга приняли положительное решение о сертификации, а сама экспертиза и расшифровка ее результатов должны занимать не более 10 рабочих дней.
На подготовку итогового отчета по результатам ее проведения исполнителю отводится 30 рабочих дней, а на его направление заявителю — 3 рабочих дня. Копия такого отчета также направляется в Минпромторг. На основании отчета формируется окончательное заключение, которое в случае положительного характера сопровождается выдачей сертификата производителю лекарственного препарата.
Документы для сертификации
Чтобы получить сертификат GMP в России, производитель обращается в уполномоченный орган с заявлением, к которому прилагает пакет документов, включающий:
- копию документа, подтверждающего наличие у заявителя полномочий по взаимодействию с контролирующей организацией;
- копия основного досье используемого производственного объекта;
- информация о фактах несоответствия препарата действующим требованиям к качеству и безопасности и о фактах отзыва медикамента из оборота за период не менее 2 лет;
- полный список лекарств, который изготавливаются на данном производственном объекте;
- копия лицензии на производство лекарств;
- письмо о согласии на проведение инспекции производства.
Важнейшие документы предоставляются заявителем в копиях, поскольку при утере их восстановить невозможно или очень сложно. Правила регламентируют, что если заявление подает иностранный производитель, и некоторые документы в составе пакета представлены на другом языке, они должны быть переведены на русский язык и заверены в установленном порядке.
Сроки сертификации
Общая продолжительность процедуры сертификации складывается из следующих сроков.
Этап сертификационной процедуры
Максимальная допустимая продолжительность
Проверка полноты пакета документации, представленной с заявлением о сертификации, и правильности ее оформления, назначение инспекции
10 рабочих дней
Направление информации о назначении инспектирования в уполномоченное учреждение, которое проводит проверку
Инспектирование и анализ лекарственного средства
160 рабочих дней
Принятие решения о выдаче заключения по результатам инспекционного отчета
10 рабочих дней
160-дневный период инспектирования включает внесение производителя в график инспекций, ожидание процедуры и проведение самой инспекции. Она должна занимать не более 10 рабочих дней.
Такой порядок действует, если в документации, поданной производителем, не обнаружат ошибок и недочетов, из-за которых ее могут направить на доработку. В этом случае вся процедура займет немногим более 180 рабочих дней, то есть свыше 8 месяцев.
Стоимость получения сертификата
Обязательной для всех производителей лекарственных средств, претендующих на получение сертификата, подтверждающего соответствие их продукции стандартам GMP, является оплата государственной пошлины за рассмотрение соответствующего заявления в Министерстве промышленности и торговли. Ее размер составляет 7500 рублей. Оплатить данную сумму необходимо еще до подачи заявления в ведомство, а ее размер никак не зависит от результатов рассмотрения документа.
Однако данная пошлина — это далеко не единственный и не самый крупный платеж, который потребуется осуществить производителю лекарств. Другой значительной статьей расходов станет плата за проведение экспертной оценки производства и продукции заявителя. Такая процедура выполняется специалистами ФГБУ «ГИЛС и НП»: для каждого из них предварительно проводится аттестация эксперта по GMP в России.
При этом размер платы за проведение оценки не является строго установленным, а определяется в зависимости от объема, характера и сложности необходимых процедур в соответствии с положениями приказа Министерства промышленности и торговли Российской Федерации от 11.01.2016 № 9 «Об утверждении методики определения размера платы за оказание услуги по инспектированию GMP». В случае, если проверка потребует проведения значительного объема работы и привлечения большого количества высококвалифицированных экспертов, размер платы за ее проведение может превышать 2,5 миллиона рублей.
Источник


