Разработка лекарств — Drug development
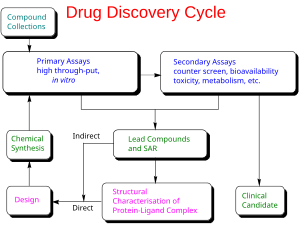
Разработка лекарств — это процесс вывода на рынок нового фармацевтического препарата после того, как в процессе открытия лекарственного средства было идентифицировано ведущее соединение . Он включает в себя доклинические исследования микроорганизмов и животных, регистрацию нормативного статуса, например, через Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США для исследования нового лекарственного препарата для начала клинических испытаний на людях, и может включать этап получения разрешения регулирующих органов с подачей заявки на новый лекарственный препарат. продавать препарат. Весь процесс — от концепции до доклинических испытаний в лаборатории до разработки клинических испытаний, включая испытания фаз I – III — до одобренной вакцины или препарата, обычно занимает более десяти лет.
СОДЕРЖАНИЕ
Разработка нового химического предприятия
В общих чертах процесс разработки лекарств можно разделить на доклиническую и клиническую работу.

Доклинические
Новые химические образования (NCE, также известные как новые молекулярные образования или NME) — это соединения, которые возникают в процессе открытия лекарств . Они обладают многообещающей активностью против конкретной биологической мишени, которая важна при заболевании. Однако мало что известно о безопасности, токсичности , фармакокинетике и метаболизме этого NCE у человека. Задача разработки лекарств — оценить все эти параметры до клинических испытаний на людях. Еще одна важная цель разработки лекарств состоит в том, чтобы рекомендовать дозу и график для первого использования в клинических испытаниях на людях (« первая доза на людях » [FIH] или первая доза для человека [FHD], ранее также известная как «первая доза для человека »). -человек «[ФИМ]).
Кроме того, при разработке лекарств необходимо установить физико-химические свойства NCE: его химический состав, стабильность и растворимость. Производители должны оптимизировать процесс, который они используют для производства химического вещества, чтобы они могли масштабироваться от фармацевтического химика, производящего миллиграммы, до производства в килограммах и тоннах . Они дополнительно исследуют продукт на пригодность для упаковки в виде капсул , таблеток , аэрозолей, внутримышечных инъекций, подкожных инъекций или внутривенных препаратов . Вместе эти процессы известны в доклинических и клинических разработках как химия, производство и контроль (CMC).
Многие аспекты разработки лекарств сосредоточены на удовлетворении нормативных требований к применению нового лекарственного средства . Как правило, они представляют собой ряд тестов, предназначенных для определения основных токсичностей нового соединения перед его первым применением на людях. Законодательство требует проведения оценки токсичности для основных органов (воздействие на сердце и легкие, мозг, почки, печень и пищеварительную систему), а также воздействия на другие части тела, на которые может повлиять препарат ( например, кожа, если новое лекарство должно быть доставлено на кожу или через нее). Такие предварительные тесты проводятся с использованием методов in vitro (например, с изолированными клетками), но во многих тестах могут использоваться только экспериментальные животные, чтобы продемонстрировать сложное взаимодействие метаболизма и воздействия лекарственного средства на токсичность.
Информация собирается из этого доклинического тестирования, а также информация о CMC и передается в регулирующие органы (в США, в FDA ) в качестве заявки на новый исследуемый препарат (IND). Если IND одобрен, разработка переходит в клиническую фазу.
Клиническая фаза
Клинические испытания включают три или четыре этапа:
- Исследования фазы I, обычно на здоровых добровольцах, определяют безопасность и дозировку.
- Испытания фазы II используются для получения первоначального представления об эффективности и дальнейшего изучения безопасности у небольшого числа пациентов, страдающих заболеванием, на которое нацелена NCE.
- Испытания фазы III — это крупные решающие испытания для определения безопасности и эффективности у достаточно большого числа пациентов с целевым заболеванием. Если безопасность и эффективность будут надлежащим образом доказаны, клинические испытания могут остановиться на этом этапе, и NCE перейдет к этапу подачи заявки на новое лекарство (NDA).
- Испытания фазы IV — это испытания после утверждения, которые иногда являются условием, устанавливаемым FDA, также называемыми исследованиями послепродажного наблюдения.
Процесс определения характеристик лекарственного средства не прекращается после того, как NCE переходит в клинические испытания на людях. Помимо тестов, необходимых для подачи новой вакцины или противовирусного препарата в клинику впервые, производители должны гарантировать, что любые долгосрочные или хронические токсические эффекты четко определены, включая воздействие на системы, которые ранее не отслеживались (фертильность, репродуктивность, иммунная система, среди прочего).
Если вакцина-кандидат или противовирусное соединение выявляется в результате этих испытаний с приемлемым профилем токсичности и безопасности, и производитель может дополнительно продемонстрировать, что он оказывает желаемый эффект в клинических испытаниях, тогда портфель доказательств NCE может быть представлен для утверждения на рынке в различных странах. где производитель планирует его продавать. В Соединенных Штатах этот процесс называется « заявка на новое лекарство » или NDA.
Большинство новых лекарственных препаратов-кандидатов (НКЭ) терпят неудачу во время разработки лекарств либо потому, что они обладают неприемлемой токсичностью, либо потому, что они просто не доказывают свою эффективность в отношении целевого заболевания, как показано в клинических испытаниях фазы II – III. Критические обзоры программ разработки лекарственных средств показывают, что клинические испытания фазы II – III терпят неудачу в основном из-за неизвестных токсических побочных эффектов (50% неудач кардиологических исследований фазы II ), а также из-за неадекватного финансирования, недостатков дизайна испытаний или плохого выполнения испытаний.
Исследование, охватывающее клинические исследования 1980–90-х годов, показало, что только 21,5% кандидатов в лекарственные препараты, которые начали испытания фазы I, в конечном итоге были одобрены для продажи. В течение 2006–15 годов показатель успешности получения одобрения от фазы I для успешных испытаний фазы III в среднем составлял менее 10%, а конкретно для вакцин — 16%. Высокий процент неудач, связанный с разработкой фармацевтических препаратов, называется «показателем истощения», требующим принятия решений на ранних стадиях разработки лекарств, чтобы «закрыть» проекты на раннем этапе, чтобы избежать дорогостоящих неудач.
Расходы
В одном исследовании 2010 года капитализированные и наличные затраты на вывод на рынок одного нового препарата оценивались примерно в 1,8 млрд долларов США и 870 млн долларов США соответственно. Медиана смета расходов 2015-16 гг исследований для разработки 10 противораковых препаратов была 648000000 $. В 2017 году средняя стоимость основного исследования по всем клиническим показаниям составила 19 миллионов долларов.
Средняя стоимость (в долларах 2013 г.) каждого этапа клинических исследований составила 25 миллионов долларов США для исследования безопасности фазы I, 59 миллионов долларов США для рандомизированного контролируемого исследования эффективности фазы II и 255 миллионов долларов США для основного исследования фазы III, чтобы продемонстрировать его эквивалентность или превосходство. к существующему одобренному препарату, возможно, до 345 миллионов долларов. Средняя стоимость проведения основного исследования фазы III на 2015–2016 гг. На кандидате от инфекционного заболевания составила 22 миллиона долларов.
Полная стоимость вывода на рынок нового лекарства (т. Е. Нового химического соединения ) — от открытия через клинические испытания до утверждения — сложна и спорна. В обзоре в 2016 году 106 кандидатов в лекарственные препараты, оцененных в ходе клинических испытаний, общие капитальные затраты для производителя, имеющего препарат, одобренный в результате успешных испытаний фазы III, составили 2,6 миллиарда долларов (в долларах 2013 года), и эта сумма увеличивается ежегодно на 8,5%. В течение 2003–2013 гг. Для компаний, которые одобрили 8–13 препаратов, стоимость одного препарата может вырасти до 5,5 млрд долларов, в основном из-за международной географической экспансии маркетинга и текущих затрат на исследования фазы IV для непрерывного наблюдения за безопасностью .
Альтернативы разработке традиционных лекарств имеют целью объединить университеты, правительства и фармацевтическую промышленность и оптимизировать ресурсы.
Оценка
Природа проекта разработки лекарств характеризуется высоким уровнем выбытия , большими капитальными затратами и длительными сроками. Это делает оценку таких проектов и компаний сложной задачей. Не все методы оценки могут справиться с этими особенностями. Наиболее часто используемые методы оценки — это чистая приведенная стоимость с поправкой на риск (rNPV), деревья решений , реальные опционы или сопоставимые активы .
Наиболее важными факторами ценности являются стоимость капитала или используемая ставка дисконтирования, атрибуты фазы, такие как продолжительность, процент успеха и затраты, а также прогнозируемые продажи, включая стоимость товаров, а также расходы на маркетинг и продажи. Менее объективные аспекты, такие как качество управления или новизна технологии, должны быть отражены в оценке денежных потоков .
Шанс успеха
Теоретически кандидаты на создание нового лекарства для лечения болезни могут включать от 5 000 до 10 000 химических соединений. В среднем около 250 из них достаточно перспективны для дальнейшей оценки с использованием лабораторных тестов, мышей и других подопытных животных. Обычно около десяти из них подходят для испытаний на людях. Исследование, проведенное Центром Тафтса по изучению разработки лекарственных средств, охватывающее 1980-е и 1990-е годы, показало, что только 21,5 процента лекарств, которые начали испытания фазы I, были в конечном итоге одобрены для продажи. За период с 2006 по 2015 год показатель успешности составил 9,6%. Высокая частота неудач, связанная с разработкой фармацевтических препаратов, называется проблемой «скорости истощения». Чтобы избежать дорогостоящих неудач, важно принимать осторожные решения во время разработки лекарств. Во многих случаях разумная программа и дизайн клинических испытаний могут предотвратить ложноотрицательные результаты. Хорошо спланированные исследования по подбору доз и сравнения как с плацебо, так и с группой лечения золотым стандартом играют важную роль в получении надежных данных.
Вычислительные инициативы
Новые инициативы включают партнерство между правительственными организациями и промышленностью, например, Европейская инициатива в области инновационных лекарственных средств . США пищевых продукты и медикаменты создали «Critical Path Initiative» для повышения инновационной разработки лекарственных средств, а также прорыв терапии назначение ускорить разработку и регулирующий обзор потенциальных лекарственных средств , для которых предварительных клинических доказательств показывает , кандидат препарат может существенно улучшить терапию для серьезного беспорядок.
В марте 2020 года Министерство энергетики США , Национальный научный фонд , НАСА , промышленность и девять университетов объединили ресурсы для доступа к суперкомпьютерам IBM в сочетании с ресурсами облачных вычислений от Hewlett Packard Enterprise , Amazon , Microsoft и Google для открытия лекарств. . Консорциум высокопроизводительных вычислений COVID ‑ 19 также нацелен на прогнозирование распространения заболеваний, моделирование возможных вакцин и скрининг тысяч химических соединений для разработки вакцины или терапии COVID ‑ 19. В мае 2020 года было запущено партнерство OpenPandemics — COVID ‑ 19 между Scripps Research и IBM World Community Grid . Партнерство представляет собой проект распределенных вычислений, который «автоматически запустит смоделированный эксперимент в фоновом режиме [подключенных домашних компьютеров], который поможет предсказать эффективность конкретного химического соединения в качестве возможного лечения COVID ‑ 19».
Источник
Разработка лекарственных средств определение
Разработка лекарственного средства начинается с синтеза новых химических соединений. Вещества со сложной структурой можно получить из различных иа очников, например растений (сердечные гликозиды), тканей животных (гепарин), микробных культур (бензилпенициллин), культур человеческих клеток (урокиназа), или посредством генно-инженерных технологий (человеческий инсулин).
Чем больше ясности во взаимоотношениях структуры и активности, тем более направленным оказывается поиск новых веществ.
а) Доклинические испытания дают информацию о биологических эффектах новых веществ. Начальный скрининг может включать биохимические и фармакологические исследования (анализ связывания с рецептором) или эксперименты на культурах клеток, изолированных клетках и изолированных органах.
Поскольку эти модели не способны воспроизвести сложные биологические процессы, происходящие в интактных организмах, любое потенциальное лекарственное средство необходимо проверить на животных. Только эксперименты на животных позволяют выяснить, возникаютли желаемые эффекты при дозах, не вызывающих токсичности или сопровождающихся слабой токсичностью. Цель токсикологических исследований заключается в том, чтобы оценить:
1) токсичность, обусловленную кратковременным или длительным приемом;
2) генетические повреждения (генотоксичность, мутагенез);
3) развитие опухолей (канцерогенность);
4) возникновение врожденных дефектов (тератогенность).
В экспериментах на животных также оценивают всасывание, распределение, метаболизм и элиминацию (фармакокинетика) изучаемых веществ. На уровне доклинического изучение лишь у малой части новых веществ обнаруживается потенциал для применения у человека.
Фармацевтическиетехнологии предлагают методы изготовления лекарственных форм.
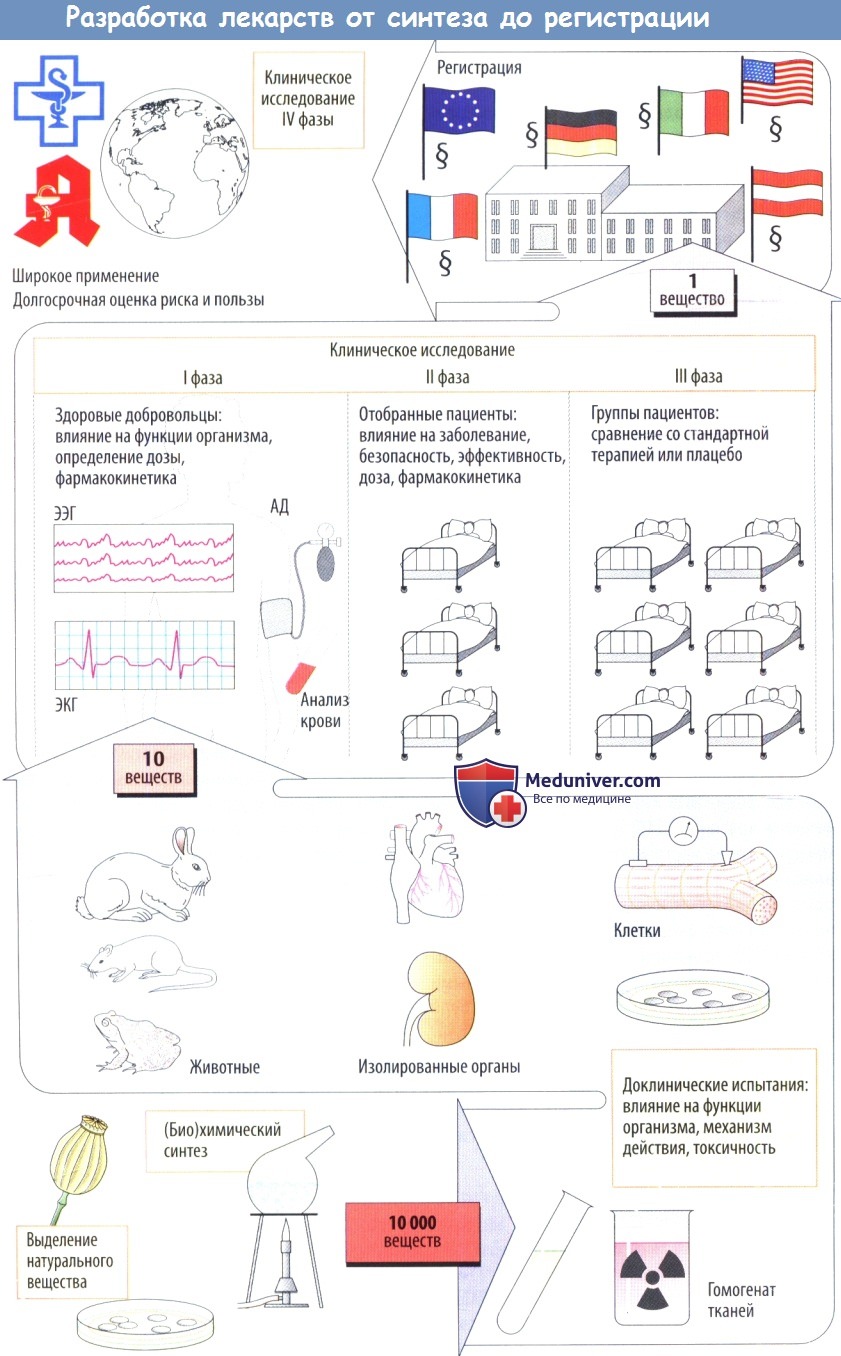
б) Клинические испытания начинаются с исследований I фазы, в которых участвуют здоровые лица; цель этих исследований — определить, будут ли эффекты, наблюдаемые у животных, также возникать у людей. Кроме того, на данном этапе определяется дозозависимость клинических эффектов.
Во II фазе потенциальные лекарственные средства сначала проверяют на отобранных пациентах на терапевтическую эффективность при заболевании, для лечения которого эти препараты предназначались. Если полезное действие очевидно, а частота побочных эффектов приемлема, начинается III фаза, в которой участвует более крупная группа пациентов, у которых новое средство сравнивают с традиционными методами лечения сточки зрения терапевтического результата.
Как форма экспериментов на людях, эти клинические исследования подвергаются анализу и одобрению этическими комитетами медицинских учреждений в соответствии с международными правилами проведения (Хельсинкской, Токийской и Венецианской декларациями). Во время клинических исследований выясняется, что многие вещества нельзя использовать. Как правило, в конце концов примерно из 10 000 вновь синтезированных веществ остается только одно.
в) Решение зарегистрировать новое лекарственное средство выносится национальным регуляторным органом (Food and Drug Administration в США, Health Protection Branch Drugs Directorate в Канаде, комиссией ЕС вместе с European Medicines Agency в Великобритании), которому производители должны подавать регистрационные документы.
Заявители должны документально подтвердить результатами соответствующих испытаний (доклинических и клинических), что критерии эффективности и безопасности удовлетворены и что лекарственные формы продукта (таблетки, капсулы и т. д.) соответствуют всем стандартам контроля качества.
После регистрации новое лекарственное средство может продаваться под торговым названием, оно должно быть доступным, выписываться врачами и отпускаться фармацевтами. В это время наблюдение продолжается в форме постмаркетинговых исследований (IV фаза клинических исследований)
г) Фармакологический надзор — действия, направленные на то, чтобы выявлять и устранять связанные с препаратом риски во время проведения клинических исследований и последующего его выхода на рынок. Фармаконадзор включает отчеты о предполагаемых случаях нежелательных реакций, направляемые в национальные регуляторные органы.
На основе длительного опыта применения можно правильно оценить соотношение риска и пользы и, следовательно, терапевтическую ценность нового лекарственного средства. Если новый препарат имеет небольшое преимущество перед существующими, необходимо принимать во внимание соотношение затрат и пользы от применения лекарственного средства.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Источник


