- Этапы разработки лекарственных препаратов
- Этапы разработки лекарственных средств [ править | править код ]
- Этапы разработки лекарственных средств
- Содержание
- Контроль качества и этапы разработки лекарственных средств [ править | править код ]
- Контроль качества лекарственных средств [ править | править код ]
- Этапы разработки лекарственных средств [ править | править код ]
Этапы разработки лекарственных препаратов
Этапы разработки лекарственных средств [ править | править код ]
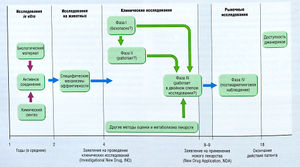
Между моментами создания нового лекарства и демонстрации его клинической эффективности и адекватной безопасности можно выделить несколько этапов (рис. 3.1). Этап первоначальной разработки обычно состоит в определении терапевтической дели (заболевание или состояние) или целевой молекулы, например рецептора, фермента и др., и последующем обнаружении основного химического соединения, т.е. вещества с характерным эффектом, необходимым для нового лекарства. В современных программах разработки лекарств чаще определяется целевая молекула, которая является ключевым звеном патологического процесса, и осуществляется поиск синтетических и природных соединений, действующих на эту молекулу. В дальнейшем пытаются разработать более подходящие соединения. Получение таких соединений — многократный процесс, включающий синтез похожих химических производных основного соединения. При разработке новых аналогов, чтобы получить требуемую эффективность, используют анализ взаимосвязи структура-активность (SAR или QSAR при количественной оценке).
Описание к рис. 3.1 Процесс разработки и оценки лекарства для вывода на рынок США. Некоторые требования для препаратов, используемых для лечения жизнеугрожающих заболеваний, могут отличаться [Katzung ВС. Basic and Clinical Pharmacology, 6th ed. New York: I Appleton & Lange]
Некоторые аналоги препаратов становятся объектами крупномасштабных фармакологических и токсикологических исследований для определения характеристик лекарств, которые могут получить одобрение для клинических исследований с участием пациентов. После серии клинических наблюдений полученные данные подаются в регулирующий орган для получения разрешения на реализацию нового лекарства. После этого с помощью различных методов собирают результаты клинического применения препарата. Этот процесс называют постмаркетинговыми наблюдениями (см. Принятие фармакотерапевтического решения), которые регулируют менее строго, чем процедуры, необходимые до получения регистрации.
Эксперименты на животных обеспечивают основу клинических наблюдений
Сведения о фармакологических эффектах лекарства in vitro и in vivo используют для предварительного заключения о его терапевтической ценности. Эти данные нужны для обоснования исследований на людях, поскольку без них не будет базы для оценки ожидаемой пользы и приемлемого риска нежелательных эффектов. Доклиническими исследованиями называют эксперименты in vitro и на животных, используемые для определения действия лекарства на уровне молекулы, клетки, определенной ткани или органа, оценки фармакологических свойств и изучения потенциальных терапевтических эффектов на животных моделях заболеваний человека. Исследования на животных также помогают изучить метаболизм и распределение лекарства в организме и разработать основные показания. Клинические исследования не могут быть продолжены, если не доказана безопасность лекарства. Для оценки возможной токсичности нового лекарства необходимы следующие исследования на животных:
- токсикологические исследования in vitro для оценки генетической и биохимической токсичности;
- оценка острой токсичности с изучением физиологических систем (сердечно-сосудистая, центральная нервная, желудочно-кишечный тракт), кожи и слизистых (острое раздражение и возбуждение);
- оценка подострой и хронической токсичности;
- оценка канцерогенности;
- оценка репродуктивной токсичности;
- оценка генетической токсичности.
При изучении острой токсичности оценивают эффекты, возникающие через несколько часов или дней после однократного введения. При изучении хронической токсичности рассматривают эффекты после введения повторных доз в течение нескольких недель или месяцев.
Однако надежность данных, полученных на животных, для прогнозирования клинических результатов зависит от уровня клинической релевантности модели. Например, модель пневмонии, вызванной золотистым стафилококком, хорошо прогнозируема. Инфицирование организма одинаково и у людей, и у животных. Иммунологический ответ против бактерий и легочная патология у животных и человека очень схожи. Напротив, животные модели других заболеваний только косвенно имитируют заболевания человека и менее предсказуемы. Обычно возможность разработки животной модели связана с пониманием патофизиологии конкретного заболевания. В указанном примере непосредственная причина пневмонии хорошо известна, в то время как точная этиология многих заболеваний не определена.
Изучение лекарства в клинике состоит из нескольких этапов
Клинические исследования начинаются после того, как собрано достаточное количество данных после исследований на животных в качестве обоснования для оценки нового лекарства в клинике и получения необходимого официального разрешения. Этапы разработки лекарства обозначают как фаза I, фаза II и фаза III. Фаза IV является этапом пост-маркетинговых наблюдений и других пострегистрационных клинических исследований (см. рис. 3.1).
Фаза I включает первые клинические исследования с участием людей. Эти исследования проводят под очень строгим наблюдением, обычно они являются открытыми или одинарными слепыми (табл.3.2) и определяют наименьшую допустимую дозу по токсичности. Дальнейшие исследования проводят с меньшими дозами. Обычно в таких исследованиях участвуют молодые здоровые мужчины. В дальнейшем их заменяют группой больных. Также в эту фазу получают первичные данные о фармакокинетике.
Фаза II начинается после определения диапазона допустимых доз и рассматривается как доказательство концепции. Этот этап проходит с участием больных, у которых новое лекарство должно проявить свой потенциальный эффект. Основная цель состоит в получении доказательств того, что новое лекарство эффективно, т.е. обладает эффектами, полученными в доклинических исследованиях. Иногда конечной точкой клинических наблюдений фазы II является собственно терапия, в других случаях используют заместительные конечные точки исследований. Заместительная конечная точка прогнозирует или предположительно прогнозирует истинную конечную точку. Например, изучение лекарства при сердечной недостаточности может иметь истинную конечную точку при увеличении толерантности к нагрузке или выживаемости. Заместительная конечная точка для того же лекарства может быть уменьшением периферического сопротивления сосудов и улучшением сердечного выброса. Для лекарства, которое может предотвращать тромбообразование при ангиопластике, заместительной конечной точкой может быть ингибирование агрегации тромбоцитов, а истинной конечной точкой — уменьшение рестеноза.
Заместительная конечная точка наиболее удобна, когда она тесно связана с истинной конечной точкой. Так, например, заместительной конечной точкой является снижение артериального давления. Целью лечения гипертензии является снижение неблагоприятных сердечно-сосудистых реакций организма и почечной недостаточности как последствий гипертензии. Таким образом, снижение артериального давления — это заместительная конечная точка для уменьшения последствий гипертензии.
Другие цели фазы II состоят в определении фармакокинетики лекарства и связи между эффектом и концентрацией вещества в плазме, если это возможно. Также изучается влияние заболеваний печени и почек на выведение лекарства из организма, фармакокинетические и фармакодинамические взаимодействия нового лекарства с другими средствами, с которыми их могут назначать совместно.
Исследования в фазу II могут быть одинарными или двойными слепыми, параллельными или перекрестными, с использованием случайных выборок пациентов. В этнически разнородных популяциях, например в США, в фармакокинетических исследованиях иногда изучают особенности метаболизма лекарств у разных этнических групп. Этническая однородность является грубым усреднением генетической классификации. Возможно, в будущем более корректный подход к оценке путей метаболизма и клинических результатов будет состоять в классификации пациентов по их генетической предрасположенности к метаболизму лекарств. Тогда будет возможно предсказать, для какого генотипа лекарство будет более полезно, а для какого — токсично. Этот раздел фармакологии называют фармакогенетикой.
В фазу III устанавливают эффективность и безопасность нового лекарства. Если возможно, проводят контролируемые рандомизированные двойные слепые исследования, которые всегда параллельны. Планируемая модель и размер всех клинических наблюдений, особенно фазы III, основывают на статистических действиях, например рандомизации процедур, чтобы после окончания исследования получить веское заключение. Кроме того, популяционные исследования фазы III должны усреднять целевую популяцию для данного лекарства. В исследовании должны участвовать пациенты с различными проявлениями изучаемого заболевания. Распределение по этническим группам и полу должно отражать таковое в популяции. Наибольшее внимание уделяют изучению детей, за исключением случаев, когда это нецелесообразно, например при изучении лекарств для лечения таких заболеваний у пожилых, как болезнь Альцгеймера.
Разработка лекарств является длительным процессом
- Время от подачи заявки на регистрацию до его получения составляет от 6 мес до нескольких лет, чаще 1-2 года
- Процесс разработки лекарства до регистрации обычно занимает 6-10 лет
Таблица 3.2 Клинические исследования, терминология
Стандартная терапия (или плацебо при отсутствии стандартов), с которой сравнивают эффективность нового препарата
Пациенты, участвующие в исследовании, имеют одинаковую возможность быть включенными в опытную или контрольную группу, а факторы, которые могут повлиять на результаты, одинаково распределены между двумя группами
Двойное слепое исследование
Ни врач, ни пациент не знают, получает ли данный пациент опытное или контрольное средство, что помогает избежать субъективизма
Одинарное слепое исследование
Врач знает, какой препарат назначен данному пациенту, но пациент не знает
Противоположно двойному слепому: и врач, и пациент знают, какое средство (опытное или контрольное)назначено и в какой дозе
Одновременно оценивают как минимум две схемы, но пациенту назначают только один вид терапии
Пациенты получают каждый вид лечения последовательно и таким образом выступают в качестве контрольной группы для самих себя. Например, если лечение А оценивают относительно лечения В, то некоторые пациенты получают сначала А, потом В, а другие наоборот — сначала В, потом А. Так оценивают эффекты лекарственной терапии, а не порядка назначений
Измеряют для оценки эффекта лекарства (например, нормализация артериального давления — конечная точка для оценки антигипертензивных средств, уменьшение боли — конечная точка для оценки анальгетиков)
Заместительная конечная точка
Результат лечения, который прогнозирует истинную цель терапии, не являясь этой целью (например, снижение размера опухоли в качестве заместителя выживаемости)
Источник
Этапы разработки лекарственных средств
Содержание
Контроль качества и этапы разработки лекарственных средств [ править | править код ]
Контроль качества лекарственных средств [ править | править код ]
В США, как и в других странах, принимаются все новые законы с целью гарантировать эффективность и безопасность выпускаемых лекарственных средств. Первый из них — Федеральное положение о пищевых продуктах и лекарственных средствах от 1906 г. — был направлен на пресечение ввоза фальсифицированных и некачественных пищевых продуктов и лекарственных средств из других стран. Однако в то время еще не было нормативов для оценки их эффективности и безопасности. После того как в 1938 г. поступивший в продажу раствор сульфаниламида в ди-этиленгликоле (очень токсичный растворитель) привел к смерти 105 детей, было принято дополнение к Федеральному положению о пищевых продуктах и лекарственных средствах. Согласно этому дополнению, ответственность за проверку безопасности и показаний к применению лекарственных средств возлагалась на ФДА. От фирм-производителей стали требовать проведения испытаний с целью выявления побочных эффектов; кроме того, теперь производители должны были получать разрешение на выпуск нового препарата. Однако в то время еще не требовались доказательства эффективности препарата и показания к применению часто не соответствовали действительности (Wax, 1995).
Между тем в промышленных и научных лабораториях велись активные исследования в области фундаментальной и клинической фармакологии, что привело к созданию множества новых препаратов. Однако поскольку четких критериев эффективности не было, показания к применению лекарственных средств не подкреплялись фактическими данными. Соотношение риска и пользы учитывалось очень редко. Однако в начале 1960-х гг. ситуация резко изменилась. Вскоре после того, как в Европе поступил в продажу талидомид (снотворное средство, не обладавшее какими-либо явными преимуществами перед другими сходными препаратами), участились случаи относительно редкого врожденного нарушения — фокомелии. Через некоторое время заболеваемость фокомелией достигла внушительных цифр, и ретроспективные эпидемиологические исследования убедительно доказали, что ее причина — прием талидомида на ранних сроках беременности. Это доказательство тератогенности, в общем, не такого уж нужного препарата потрясло весь мир, и в 1962 г. в США были приняты Поправки Кифовера—Харриса к Положению о пищевых продуктах, лекарственных и косметических средствах.
Поправки Кифовера—Харриса имеют силу закона и содержат требования к проведению фармакологических и токсикологических исследований у животных на доклиническом этапе изучения лекарственных средств. Полученные результаты представляют в ФДА в виде заявки на клинические испытания препарата. После этого проводят клинические испытания, включающие три фазы (см. ниже), и на основании полученных данных ФДА выдает разрешение на выпуск препарата. Для всех препаратов, поступивших в продажу после 1962 г., требуются доказательства эффективности и данные о соотношении риска и пользы. В Поправках Кифовера—Харриса содержатся также требования к фирмам-производителям предоставить сведения об эффективности всех препаратов, выпущенных с 1938 по 1962 г.
Эффективность лекарственных средств оценивают в «адекватных и контролируемых испытаниях». Обычно проводят два клинических испытания, которые, как правило, являются рандомизированными двойными слепыми плацебо-контролируемыми. К моменту подачи документов на разрешение препарата база данных должна быть достаточно большой для того, чтобы судить о его безопасности. Ужесточение требований ФДА привело к тому, что увеличилось число клинических испытаний и участвующих в них добровольцев, возросли стоимость разработки препарата и время, необходимое для получения разрешения. Кроме того, рассмотрение большего количества документов стало отнимать больше времени, и в 1990 г. на него уходило уже около 3 лет. В результате усилилось противостояние между ФДА, призванным защищать общественное здоровье, и фирма-ми-производителями, желающими быстрее выпускать новые лекарственные средства и получать прибыль от их продажи. Мнение общественности разделилось: одни критиковали ФДА за затягивание этого процесса, другие же — за то, что ряд побочных эффектов выявляют уже после поступления препаратов в продажу. Перед ФДА была поставлена сложная задача — тщательно проверять данные о безопасности и об эффективности лекарственных средств, но своевременно выдавать разрешения на их выпуск.
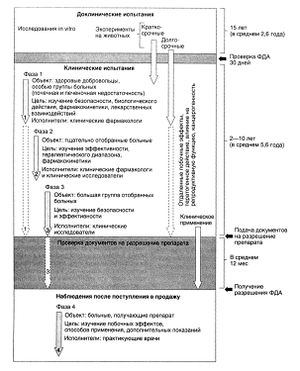
Начиная с конца 1980-х гт. под давлением обществ по борьбе со СПИДом ФДА предприняло ряд мер, чтобы упростить получение разрешения. И если раньше новые лекарственные средства появлялись в США гораздо позднее, чем в других странах, то теперь эту проблему удалось решить (Kessler et al., 1996). Во-первых, больные с угрожающими жизни заболеваниями могут принимать препарат еше до получения разрешения, если есть какие-либо данные о его эффективности и не выявлено тяжелых побочных эффектов, а имеющиеся методы лечения неэффективны (рис. 3.5). Во-вторых, ускорено рассмотрение документов на лекарственные средства, которые предполагается использовать в лечении таких заболеваний. Конгресс принял Закон об оплате сертификации лекарственных средств, согласно которому фирма-производитель должна заплатить за сертификацию своего препарата. Эти деньги пошли на то, чтобы увеличить штат ФДА; в результате рассмотрение документов ускорилось (Shulman and Kaitin, 1996). Документы на препараты, относящиеся к новым классам либо используемые в лечении опасных заболеваний, рассматриваются в первую очередь. Наконец, сотрудничая с фирмами-производителями в процессе разработки лекарственных средств, ФДА помогает уменьшить время с момента подачи заявки на клинические испытания до получения разрешения. Взаимодействие касается и планирования прицельных клинических испытаний, в которых используют либо косвенные критерии эффективности, либо прямые критерии, но не выживаемость и не частоту осложнений. Это позволяет сократить сроки получения данных, чтобы раньше оценить соотношение риска и пользы и получить разрешение на выпуск препарата. В некоторых случаях испытания 3-й фазы сокращаются или становятся вовсе не нужными. Однако при этом иногда остаются нерешенные вопросы, касающиеся безопасности, эффективности и рационального применения нового препарата. В таких случаях его могут направлять для изучения в отдельные клиники и проводить дополнительные испытания после поступления в продажу. Если эти испытания показывают, что препарат не соответствует предъявляемым требованиям, либо выявляют высокую частоту побочных эффектов или низкую эффективность, препарат может быть снят с производства. В 1997 г. эти изменения были внесены в Положение о модернизации ФДА (Suydam and Elder,1999). Вследствие предпринятых мер на рассмотрение документов стало уходить менее 1 года, и вскоре планируется сократить сроки еще больше — до 10 мес. Подробнее с содержанием данного документа, в котором описаны и многие другие направления деятельности ФДА, можно ознакомиться в интернете по адресу www.fda.gov/opaconi/7modact.html. Главной предпосылкой к его разработке послужило желание общества принять риск, сопряженный с применением новых препаратов, когда речь идет об угрожающих жизни заболеваниях. Высказываются некоторые опасения, что чем проще будет получить разрешение, тем больше появится непроверенных лекарственных средств. Однако если при этом удастся гарантировать их безопасность, то сокращение сроков рассмотрения документов может принести пользу многим бальным, страдающим такими заболеваниями.
В Положении о пищевых продуктах, лекарственных и косметических средствах есть пункт, который на первый взгляд кажется противоречивым: ФДА не имеет отношения к клинической практике. Если однажды доказано, что новый препарат эффективен и безопасен, его можно выпускать, а затем врачи будут решать, как его лучше использовать. Однако важно помнить, что применение новых лекарственных средств сопряжено с более высоким риском, поскольку данных об их эффектах меньше. На сегодняшний день невозможно заранее выявить все возможные реакции, поэтому требуется систематическое наблюдение после поступления препарата в продажу.
К каждому лекарственному средству должна прилагаться инструкция по применению, составляемая ФДА и фирмой-производителем. В ней кратко освещены фармакологические свойства препарата, показания, противопоказания, меры предосторожности, побочные эффекты, средние дозы, выпускаемые лекарственные формы. Все рекламные материалы должны в точности соответствовать этой инструкции.
На сегодняшний день ФДА почти не контролирует выпуск пищевых добавок: витаминов, минералов, белковых препаратов, фитопрепаратов. Раньше к ним предъявлялись те же требования, что и к пищевым продуктам и лекарственным средствам (в зависимости от состава и показаний), однако в 1994 г. Конгресс принял Закон о пищевых добавках, который ограничил полномочия ФДА. Согласно этому закону, пищевые добавки — это вещества, которые служат дополнением к пище и содержат «(А) витамин; (Б) минеральное вещество; (В) растительный компонент; (Г) аминокислоту; (Д) средство для повышения калорийности пищи у мужчин; (Е) концентрат, метаболит, компонент, экстракт или комбинацию веществ, относящихся к классам (А), (Б), (В), (Г), (Д)». Разрешение ФДА на выпуск пищевых добавок не требуется, если они не предназначены для диагностики, лечения, профилактики каких-либо заболеваний. Однако многие естественные состояния — беременность, климактерический период, старение, половое созревание — не являются болезнью. Поэтому, если в показаниях перечислены приливы, предменструальный синдром, утреннее головокружение у беременных, ухудшение памяти у пожилых, алопеция, юношеские угри, то разрешение ФДА получать не нужно. Это касается также «укрепления здоровья» и других показаний, напрямую не связанных с заболеваниями (например, «это средство помогает расслабиться» или «поддерживает нормальное кровообращение»). В инструкциях ко многим пищевым добавкам, имеющим подобные показания, написано: «Эти данные не оценивались ФДА. Средство не имеет отношения к диагностике, лечению и профилактике каких-либо заболеваний». ФДА не может изъять такие пищевые добавки из продажи, пока не доказан «значительный или неоправданный риск заболевания» при их правильном использовании. Таким образом, всю ответственность за безопасность пищевых добавок несет только фирма-производитель.
После вступления в силу Закона о пищевых добавках на рынке появилось множество непроверенных средств. Уже отмечены случаи возникновения тяжелых побочных эффектов и взаимодействий пищевых добавок с лекарственными средствами (Fugh-Berman, 2000). Во многом сложившаяся ситуация напоминает отсутствие контроля над выпуском лекарственных средств, которое длилось до тех пор, пока в 1938 г. из-за применения раствора сульфаниламида в диэтиленгликоле не погибли 105 детей (см. выше). Врачи и больные должны помнить, что процесс разработки и выпуска пищевых добавок почти не контролируются. Об их побочных эффектах или предполагаемых взаимодействиях с лекарственными средствами нужно сообщать в ФДА по телефону или через интернет (см. ниже).
Этапы разработки лекарственных средств [ править | править код ]
Обычно врачей мало интересует процесс разработки лекарственных средств. И все же нужно иметь о нем некоторое представление, чтобы уметь оценить соотношение риска и пользы и понимать ограниченность данных об эффективности и о безопасности выпускаемых препаратов.
Сначала фармакокинетику, фармакодинамику и побочные эффекты препарата изучают in vitro и у нескольких видов животных в соответствии с опубликованными нормативами ФДА. Значимость многих требований, предъявляемых на доклиническом этапе исследования, бесспорна (например, выявления прямого токсического влияния на органы и описания дозозависимых эффектов), однако в других случаях она сомнительна, особенно из-за хорошо известных различий действия препаратов на разные виды животных. По результатам многих доклинических исследований нельзя точно предсказать, каковы будут эффекты препарата у человека, но, как ни парадоксально, при соблюдении мер предосторожности клинические испытания нового препарата относительно безопасны.
В США клинические испытания обычно включают три фазы (рис. 3.5), по окончании которых в ФДА подаются документы для получения разрешения на выпуск препарата. Основная цель клинических испытаний — оценить риск побочных эффектов препарата, и это гораздо сложнее, чем доказать его эффективность при каком-либо заболевании. В 3-й фазе обычно участвуют около 2000—3000 тщательно отобранных добровольцев. Как правило, из них только несколько сотен принимают новый препарат более 3—6 мес (на практике может потребоваться более длительное лечение). Следовательно, в ходе 3-й фазы можно выявить только самые тяжелые побочные эффекты, частота которых составляет более 1:100, возникающие почти сразу после начала приема. Другие значимые побочные эффекты, которые возникают не сразу или встречаются с частотой менее 1:1000, могут не выявляться. Действительно, многие неожиданные реакции на препарат обнаруживают только после его поступления в продажу. В еще большей степени это относится к препаратам, используемым у детей, так как клинических испытаний с их участием проводят мало. По этой причине во многих странах, в том числе в США, разработаны методики систематического наблюдения за эффектами препаратов после их поступления в продажу (Brewer and Colditz, 1999).
Источник


