Распределение лекарственных веществ факторы
Лекарственное средство после захвата организмом распределяется в кровь и через нее в различные ткани организма. Распределение может ограничиваться внеклеточным пространством (плазма + интерстициальное пространство) или может включать и внутриклеточное пространство.
Некоторые лекарственные средства активно связываются тканевыми структурами, поэтому их концентрации в плазме значительно падают еще до начала выделения.
После распределения в кровь макромолекулы остаются в основном в сосудистом пространстве, т. к. они не могут проникнуть через гистогематический барьер или даже фенестрированный эндотел ий капилляров. Это свойство используется при потере крови, когда необходимо восполнить объем циркулирующей крови, например, путем инфузии растворов с декстраном.
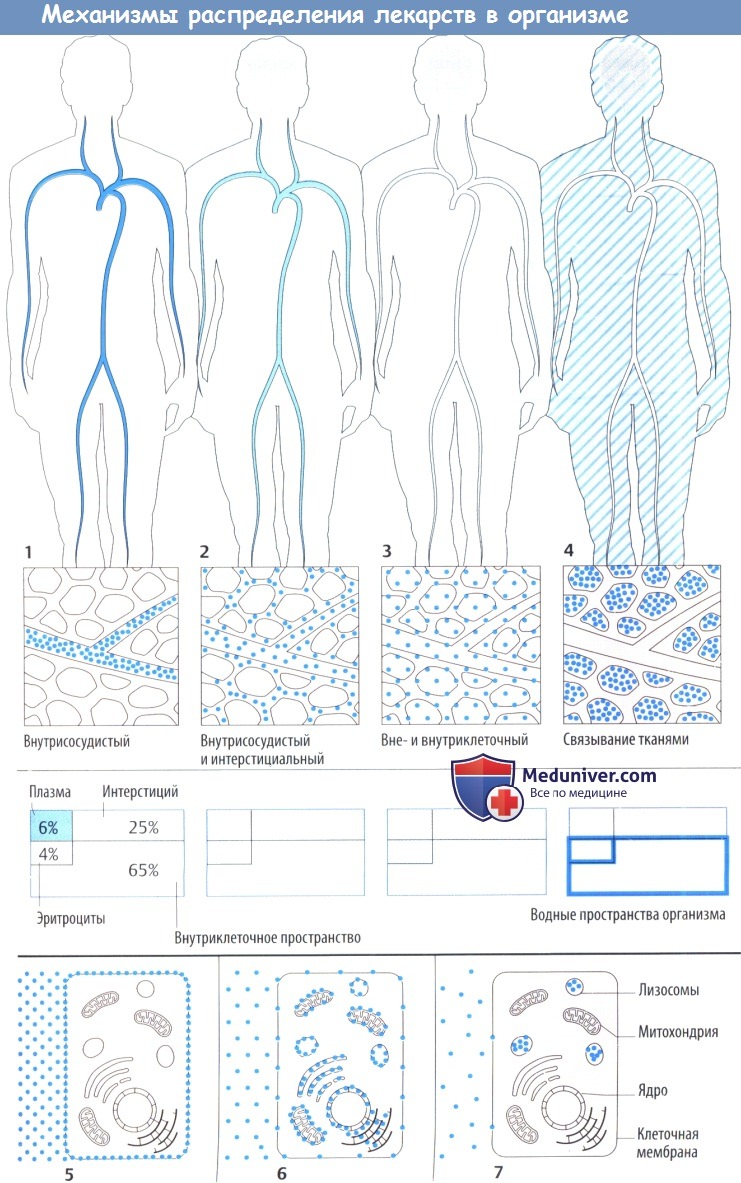
Более того, в сосудистом пространстве преимущественно находятся вещества, обладающие высоким аффинитетом к белкам плазмы (определение объема плазмы с помощью связанных с белком красителей). Несвязанное (свободное) лекарственное средство покидает кровоток, но с различной скоростью, т. к. гистогематический барьер имеет неодинаковое строение в сосудах различных органов. Эти локальные отличия не показаны на приведенных рисунках.
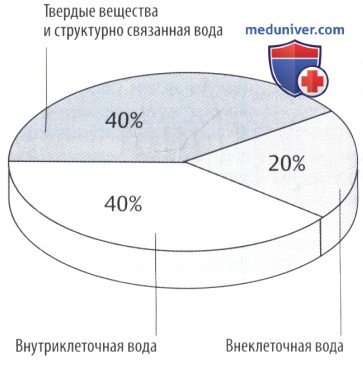
Распределение в организме определяется свойством проникать сквозь мембранные барьеры. Гидрофильные вещества (инулин) не захватываются клетками и не связываются структурами клеточной поверхности, поэтому могут использоваться для определения внеклеточного объема жидкости. Липофильные вещества диффундируют сквозь клеточную мембрану; в результате достигается равномерное распределение в жидкостях организма.
Масса тела может снизиться, как показано на секторной диаграмме. Другие подразделы указаны на рисунке ниже.
Отношение объема интерстициальной/внеклеточной жидкости варьирует в зависимости от возраста и массы тела. В процентном отношении объем интерстициальной жидкости высокий у недоношенных детей или у здоровых новорожденных (до 50% массы тела) и снижается при ожирении и старении.
Концентрация (с) раствора соответствует количеству (D) вещества, растворенного в объеме (V); из этого следует, что с = D/V. Если известна доза лекарственного препарата (D) и его концентрация в плазме (с), то можно вычислить объем распределения (V) по формуле: V=D/c. Тем не менее это кажущийся (теоретический) объем распределения (Vapp), т. к. при расчете предполагается равномерное распределение в организме.
Равномерное распределение нехарактерно при связывании лекарственных средств клеточными мембранами или мембранами внутриклеточных органелл либо при накоплении внутри органелл. В таких случаях концентрация в плазме уменьшается и Vapp может превысить фактическую величину имеющегося объема жидкости. Наоборот, если значительная часть молекул лекарственного вещества связана с белками плазмы, то с увеличивается, а вычисляемый Vapp может быть меньше, чем биологическое значение.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Источник
Распределение лекарственных препаратов по тканям
, PharmD, MAS, BCPS-ID, FIDSA, FCCP, FCSHP, Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California San Diego
После того как лекарственное средство попадает в системный кровоток, оно распределяется в тканях организма. Распределение обычно происходит неравномерно из-за различий в интенсивности кровоснабжения, связывания с тканями (например, с различным содержанием жира), местного рН и проницаемости клеточных мембран.
Степень проникновения лекарственного средства в ткань зависит от скорости кровотока, массы ткани и характера распределения вещества между кровью и тканью. Равновесное распределение (когда скорости проникновения и выхода из ткани совпадают) достигается быстрее в областях с богатой васкуляризацией, если диффузия через клеточную мембрану не является скорость-лимитирующим фактором. После достижения равновесия концентрация лекарственного средства в ткани и внеклеточных жидкостях пропорциональна концентрации в плазме крови. Метаболизм и элиминация происходят одновременно с распределением, делая процесс динамичным и сложным.
После того, как лекарственное средство проникло в ткани, его распределение в интерстициальной жидкости определяется, в первую очередь, перфузией. Для мало перфузируемых тканей (например, мышечной, жировой) характерно очень медленное распределение, особенно если ткань обладает высоким сродством к лекарственному веществу.
Объем распределения
Кажущийся объем распределения – это гипотетический объем жидкости, в котором могло бы распределиться общее количество введенного лекарственного средства для создания концентрации, соответствующей таковой в плазме крови. Например, если вводится 1 000 мг лекарственного средства, а концентрация в плазме крови составляет 10 мг/л, то 1 000 мг распределяется в 100 л (доза/объем = концентрация; 1 000 мг/ x л = 10 мг/л; отсюда: x = 1 000 мг/10мг/л = 100 л).
Объем распределения не имеет никакого отношения к объему тела или содержанию в нем жидкости, а, скорее, зависит от характера распределения лекарственного вещества в организме. В случае препаратов, интенсивно связывающихся с тканями, очень малая их доля остается в системе кровообращения. Следовательно, концентрация в плазме крови будет низкой, а объем распределения – высоким. Лекарственные средства, которые преимущественно остаются в кровотоке, обычно имеют низкий объем распределения.
Объем распределения служит эталоном для плазменной концентрации, ожидаемой для введенной дозы, но дает мало информации о конкретной схеме распределения. Каждый препарат по-своему распределяется в организме. Одни препараты распределяются в основном в жировой ткани, другие – остаются во внеклеточной жидкости, а некоторые в значительной степени связаны с конкретными тканями.
Лекарственные препараты, являющиеся слабыми кислотами (например, варфарин, аспирин), зачастую хорошо связываются с белками плазмы и поэтому имеют невысокий кажущийся объем распределения. Многие основания (например, амфетамин, петидин), напротив, в большой степени захватываются тканями и, таким образом, имеют кажущийся объем распределения больше, чем объем всего организма.
Связывание
Степень распределения ЛС в ткани зависит от его относительного связывания с белками плазмы крови и тканями. В кровотоке лекарственные средства транспортируются частично как свободная (несвязанная) фракция, а частично – как связанная фракция (например, с белками плазмы крови или клетками крови). Из множества белков плазмы, которые могут взаимодействовать с препаратами, наиболее важными являются альбумин, альфа-1 кислый гликопротеин и липопротеины. ЛС-слабые кислоты обычно более интенсивно связываются с альбумином; основания, напротив, – с альфа-1-кислым гликопротеином и/или липопротеинами.
Только несвязанное лекарственное средство способно к пассивной диффузии в экстраваскулярные пространства или ткани, где происходит его фармакологическое действие. Поэтому концентрация несвязанного лекарственного средства в системном кровотоке обычно определяет концентрацию его в месте реализации эффекта и, таким образом, выраженность последнего.
Лекарственные препараты способны связываться с различными веществами помимо белков. Связывание обычно происходит, когда лекарственное средство взаимодействует с макромолекулой в водной среде, но может также произойти, когда оно проникает в жировую ткань организма. Поскольку она слабо перфузируется, время достижения равновесного состояния обычно длительное, особенно если препарат является высоколипофильным.
Накопление лекарственных средств в тканях или компартментах организма может быть причиной пролонгирования их эффекта, т.к. ткани высвобождают накопленный препарат по мере того, как снижается концентрация его в плазме крови. Например, тиопентал обладает высокой липофильностью, быстро проникает в головной мозг после однократного внутривенного введения и характеризуется развитием выраженного и быстрого анестезирующего эффекта; затем его действие прекращается в течение нескольких минут по мере того, как он перераспределяется в медленно перфузируемую жировую ткань. Затем тиопентал медленно высвобождается из запасов жира, поддерживая субанестетическую концентрацию в плазме крови. При повторном введении концентрация может стать значительной, приводя к тому, что препарат в большом количестве накопится в жировой ткани. Таким образом, этот процесс сначала сокращает время действия лекарственного средства, а затем продлевает его.
Некоторые лекарственные средства накапливаются в клетках вследствие связывания с белками, фосфолипидами или нуклеиновыми кислотами. Например, концентрация хлорохина в лейкоцитах и гепатоцитах может быть в тысячу раз выше, чем в плазме крови. Лекарственное вещество в клетках находится в равновесии с его концентрацией в плазме крови и переходит туда по мере элиминации препарата из организма.
Гематоэнцефалический барьер
Лекарственные средства проникают в центральной нервной системы по капиллярам мозга и через спинномозговую жидкость (СМЖ). Хотя головной мозг получает примерно 1/6 сердечного выброса, распределение препаратов в ткань головного мозга ограниченно, поскольку проницаемость головного мозга отличается от других тканей. Хотя некоторые жирорастворимые лекарственные средства (например, тиопентал) легко попадают в головной мозг, проникновение полярных соединений затруднено. Причиной этого является гематоэнцефалический барьер, который состоит из эндотелия капилляров головного мозга и астроцитарных отростков. Эндотелиальные клетки капилляров головного мозга, которые более тесно соединены друг с другом, чем клетки других капилляров, замедляют диффузию водорастворимых лекарственных средств. Астроцитарная оболочка состоит из слоя глиальных клеток соединительной ткани (астроцитов), примыкающего к базальной мембране эндотелия капилляров. С возрастом защитная функция гематоэнцефалического барьера становится менее эффективной, что приводит к повышению проникновения различных веществ в головной мозг.
Лекарственные вещества могут попадать в спинномозговую жидкость желудочков через хориоидальное сплетение, затем пассивно диффундируя в ткань головного мозга из ликвора. Кроме того, в хориоидальном сплетении органические кислоты (например, пенициллин) активно транспортируются из спинномозговой жидкости в кровь.
Скорость проникновения лекарственного средства в спинномозговую жидкость, как и в случае других тканей, определяется в основном мерой связывания с белками, степенью ионизации и коэффициентом распределения лекарственного средства в жирах и воде. Проникновение в головной мозг замедлено для препаратов, в значительной степени связанных с белками, и практически отсутствует для ионизированных форм слабых кислот и оснований. Поскольку ЦНС хорошо кровоснабжается, скорость распределения лекарственного средства определяется, прежде всего, проницаемостью клеточных мембран.
Источник
Распределение лекарственных препаратов по тканям
, PharmD, MAS, BCPS-ID, FIDSA, FCCP, FCSHP, Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California San Diego
После того как лекарственное средство попадает в системный кровоток, оно распределяется в тканях организма. Распределение обычно происходит неравномерно из-за различий в интенсивности кровоснабжения, связывания с тканями (например, с различным содержанием жира), местного рН и проницаемости клеточных мембран.
Степень проникновения лекарственного средства в ткань зависит от скорости кровотока, массы ткани и характера распределения вещества между кровью и тканью. Равновесное распределение (когда скорости проникновения и выхода из ткани совпадают) достигается быстрее в областях с богатой васкуляризацией, если диффузия через клеточную мембрану не является скорость-лимитирующим фактором. После достижения равновесия концентрация лекарственного средства в ткани и внеклеточных жидкостях пропорциональна концентрации в плазме крови. Метаболизм и элиминация происходят одновременно с распределением, делая процесс динамичным и сложным.
После того, как лекарственное средство проникло в ткани, его распределение в интерстициальной жидкости определяется, в первую очередь, перфузией. Для мало перфузируемых тканей (например, мышечной, жировой) характерно очень медленное распределение, особенно если ткань обладает высоким сродством к лекарственному веществу.
Объем распределения
Кажущийся объем распределения – это гипотетический объем жидкости, в котором могло бы распределиться общее количество введенного лекарственного средства для создания концентрации, соответствующей таковой в плазме крови. Например, если вводится 1 000 мг лекарственного средства, а концентрация в плазме крови составляет 10 мг/л, то 1 000 мг распределяется в 100 л (доза/объем = концентрация; 1 000 мг/ x л = 10 мг/л; отсюда: x = 1 000 мг/10мг/л = 100 л).
Объем распределения не имеет никакого отношения к объему тела или содержанию в нем жидкости, а, скорее, зависит от характера распределения лекарственного вещества в организме. В случае препаратов, интенсивно связывающихся с тканями, очень малая их доля остается в системе кровообращения. Следовательно, концентрация в плазме крови будет низкой, а объем распределения – высоким. Лекарственные средства, которые преимущественно остаются в кровотоке, обычно имеют низкий объем распределения.
Объем распределения служит эталоном для плазменной концентрации, ожидаемой для введенной дозы, но дает мало информации о конкретной схеме распределения. Каждый препарат по-своему распределяется в организме. Одни препараты распределяются в основном в жировой ткани, другие – остаются во внеклеточной жидкости, а некоторые в значительной степени связаны с конкретными тканями.
Лекарственные препараты, являющиеся слабыми кислотами (например, варфарин, аспирин), зачастую хорошо связываются с белками плазмы и поэтому имеют невысокий кажущийся объем распределения. Многие основания (например, амфетамин, петидин), напротив, в большой степени захватываются тканями и, таким образом, имеют кажущийся объем распределения больше, чем объем всего организма.
Связывание
Степень распределения ЛС в ткани зависит от его относительного связывания с белками плазмы крови и тканями. В кровотоке лекарственные средства транспортируются частично как свободная (несвязанная) фракция, а частично – как связанная фракция (например, с белками плазмы крови или клетками крови). Из множества белков плазмы, которые могут взаимодействовать с препаратами, наиболее важными являются альбумин, альфа-1 кислый гликопротеин и липопротеины. ЛС-слабые кислоты обычно более интенсивно связываются с альбумином; основания, напротив, – с альфа-1-кислым гликопротеином и/или липопротеинами.
Только несвязанное лекарственное средство способно к пассивной диффузии в экстраваскулярные пространства или ткани, где происходит его фармакологическое действие. Поэтому концентрация несвязанного лекарственного средства в системном кровотоке обычно определяет концентрацию его в месте реализации эффекта и, таким образом, выраженность последнего.
Лекарственные препараты способны связываться с различными веществами помимо белков. Связывание обычно происходит, когда лекарственное средство взаимодействует с макромолекулой в водной среде, но может также произойти, когда оно проникает в жировую ткань организма. Поскольку она слабо перфузируется, время достижения равновесного состояния обычно длительное, особенно если препарат является высоколипофильным.
Накопление лекарственных средств в тканях или компартментах организма может быть причиной пролонгирования их эффекта, т.к. ткани высвобождают накопленный препарат по мере того, как снижается концентрация его в плазме крови. Например, тиопентал обладает высокой липофильностью, быстро проникает в головной мозг после однократного внутривенного введения и характеризуется развитием выраженного и быстрого анестезирующего эффекта; затем его действие прекращается в течение нескольких минут по мере того, как он перераспределяется в медленно перфузируемую жировую ткань. Затем тиопентал медленно высвобождается из запасов жира, поддерживая субанестетическую концентрацию в плазме крови. При повторном введении концентрация может стать значительной, приводя к тому, что препарат в большом количестве накопится в жировой ткани. Таким образом, этот процесс сначала сокращает время действия лекарственного средства, а затем продлевает его.
Некоторые лекарственные средства накапливаются в клетках вследствие связывания с белками, фосфолипидами или нуклеиновыми кислотами. Например, концентрация хлорохина в лейкоцитах и гепатоцитах может быть в тысячу раз выше, чем в плазме крови. Лекарственное вещество в клетках находится в равновесии с его концентрацией в плазме крови и переходит туда по мере элиминации препарата из организма.
Гематоэнцефалический барьер
Лекарственные средства проникают в центральной нервной системы по капиллярам мозга и через спинномозговую жидкость (СМЖ). Хотя головной мозг получает примерно 1/6 сердечного выброса, распределение препаратов в ткань головного мозга ограниченно, поскольку проницаемость головного мозга отличается от других тканей. Хотя некоторые жирорастворимые лекарственные средства (например, тиопентал) легко попадают в головной мозг, проникновение полярных соединений затруднено. Причиной этого является гематоэнцефалический барьер, который состоит из эндотелия капилляров головного мозга и астроцитарных отростков. Эндотелиальные клетки капилляров головного мозга, которые более тесно соединены друг с другом, чем клетки других капилляров, замедляют диффузию водорастворимых лекарственных средств. Астроцитарная оболочка состоит из слоя глиальных клеток соединительной ткани (астроцитов), примыкающего к базальной мембране эндотелия капилляров. С возрастом защитная функция гематоэнцефалического барьера становится менее эффективной, что приводит к повышению проникновения различных веществ в головной мозг.
Лекарственные вещества могут попадать в спинномозговую жидкость желудочков через хориоидальное сплетение, затем пассивно диффундируя в ткань головного мозга из ликвора. Кроме того, в хориоидальном сплетении органические кислоты (например, пенициллин) активно транспортируются из спинномозговой жидкости в кровь.
Скорость проникновения лекарственного средства в спинномозговую жидкость, как и в случае других тканей, определяется в основном мерой связывания с белками, степенью ионизации и коэффициентом распределения лекарственного средства в жирах и воде. Проникновение в головной мозг замедлено для препаратов, в значительной степени связанных с белками, и практически отсутствует для ионизированных форм слабых кислот и оснований. Поскольку ЦНС хорошо кровоснабжается, скорость распределения лекарственного средства определяется, прежде всего, проницаемостью клеточных мембран.
Источник


