Распределение лекарственных веществ это
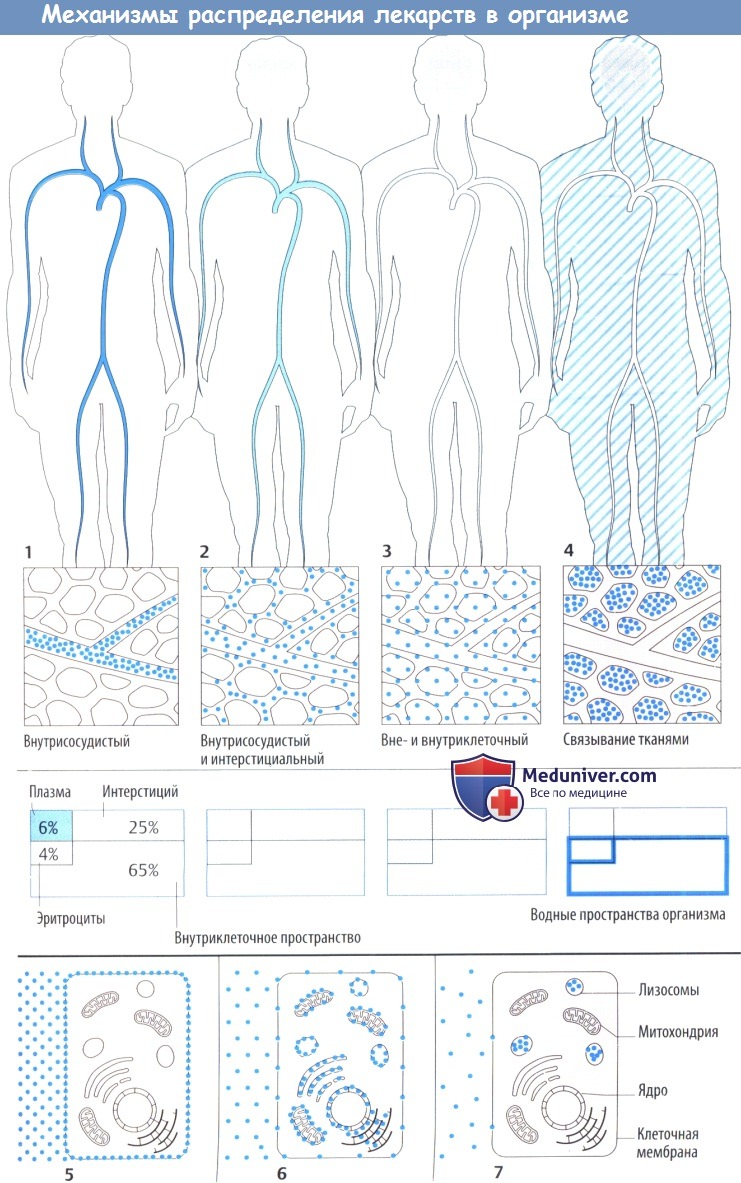
Лекарственное средство после захвата организмом распределяется в кровь и через нее в различные ткани организма. Распределение может ограничиваться внеклеточным пространством (плазма + интерстициальное пространство) или может включать и внутриклеточное пространство.
Некоторые лекарственные средства активно связываются тканевыми структурами, поэтому их концентрации в плазме значительно падают еще до начала выделения.
После распределения в кровь макромолекулы остаются в основном в сосудистом пространстве, т. к. они не могут проникнуть через гистогематический барьер или даже фенестрированный эндотел ий капилляров. Это свойство используется при потере крови, когда необходимо восполнить объем циркулирующей крови, например, путем инфузии растворов с декстраном.
Более того, в сосудистом пространстве преимущественно находятся вещества, обладающие высоким аффинитетом к белкам плазмы (определение объема плазмы с помощью связанных с белком красителей). Несвязанное (свободное) лекарственное средство покидает кровоток, но с различной скоростью, т. к. гистогематический барьер имеет неодинаковое строение в сосудах различных органов. Эти локальные отличия не показаны на приведенных рисунках.
Распределение в организме определяется свойством проникать сквозь мембранные барьеры. Гидрофильные вещества (инулин) не захватываются клетками и не связываются структурами клеточной поверхности, поэтому могут использоваться для определения внеклеточного объема жидкости. Липофильные вещества диффундируют сквозь клеточную мембрану; в результате достигается равномерное распределение в жидкостях организма.
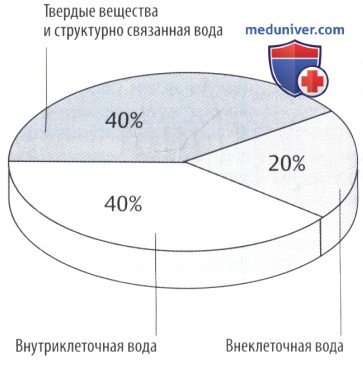
Масса тела может снизиться, как показано на секторной диаграмме. Другие подразделы указаны на рисунке ниже.
Отношение объема интерстициальной/внеклеточной жидкости варьирует в зависимости от возраста и массы тела. В процентном отношении объем интерстициальной жидкости высокий у недоношенных детей или у здоровых новорожденных (до 50% массы тела) и снижается при ожирении и старении.
Концентрация (с) раствора соответствует количеству (D) вещества, растворенного в объеме (V); из этого следует, что с = D/V. Если известна доза лекарственного препарата (D) и его концентрация в плазме (с), то можно вычислить объем распределения (V) по формуле: V=D/c. Тем не менее это кажущийся (теоретический) объем распределения (Vapp), т. к. при расчете предполагается равномерное распределение в организме.
Равномерное распределение нехарактерно при связывании лекарственных средств клеточными мембранами или мембранами внутриклеточных органелл либо при накоплении внутри органелл. В таких случаях концентрация в плазме уменьшается и Vapp может превысить фактическую величину имеющегося объема жидкости. Наоборот, если значительная часть молекул лекарственного вещества связана с белками плазмы, то с увеличивается, а вычисляемый Vapp может быть меньше, чем биологическое значение.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Источник
Распределение лекарственных веществ в организме
Распределение — это переход лекарства из системного кровотока в органы и ткани организма. Большинство ЛС распределяется неравномерно и лишь незначительная часть — относительно равномерно (некоторые ингаляционные средства для наркоза).
На характер распределения влияют многие факторы, но наиболее важными являются :
Растворимость ЛС в воде и липидах. Гидрофильные ЛС, имеющие малый молекулярный вес, легко проходят во внеклеточные области, но не могут проникнуть через мембраны клеток и (или) биологические барьеры. Липофильные ЛС легко проникают через биологические барьеры и обычно быстро распространяются по всему организму. Нерастворимые в жирах и воде ЛС могут проникать через мембраны клеток при наличии особой трансмембранной энергозависимой транспортной системы.
Степень связывания ЛС с белками. Лекарственный препарат, попав в кровь, находится в ней в двух фракциях: свободной и связанной (ЛС, связанные с белком, не взаимодействуют с рецепторами, ферментами и не проникают через клеточные мембраны). Главным образом лекарства связываются с альбуминами. Уменьшение связанной фракции лекарства на 10–20% приведет к увеличению свободной фракции на 50–100%, что важно при использовании препаратов с малой широтой терапевтического диапазона.
Особенности регионарного кровотока. Естественно, что после попадания ЛС в систему циркуляторного русла оно, в первую очередь, достигает наиболее хорошо кровоснабжаемых органов (сердце, легкие, печень, почки).
Наличие биологических барьеров, которые встречаются на пути распространения ЛС : плазматические мембраны, стенка капилляров (гистогематический барьер), ГЭБ, плацентарный барьер.
Гистогематический барьерразделяет плазму крови и интерстициальное пространство. По сравнению с другими барьерами капиллярная стенка наиболее легко проницаема для лекарств. ЛС проникают через щели, имеющиеся в местах контактов эндотелиальных клеток, выстилающих капилляры изнутри.
Липидорастворимые вещества очень быстро диффундируют через мембрану, водорастворимые и ионы — через поры.
Гематоэнцефалический барьеротносится к числу сложнейших в анатомическом и функцональном отношениях. Его проницаемость для лекарств определяет степень их центрального действия и потому представляет особый интерес для фармакологии. Собственно ГЭБ — барьер между кровью и интерстициальной жидкостью мозга. ГЭБ представлен капиллярной стенкой, диффузным основным веществом и выстилающими ее снаружи клетками и отростками нейроглии — опорной ткани мозга.
В целом ГЭБ ведет себя как типичная липидная мембрана, непроходимая для ионизированных молекул. При выраженном кислородном голодании, травматическом шоке, черепно-мозговой травме (ЧМТ), воспалении мозговых оболочек проницаемость ГЭБ для лекарств вообще и тех, что обычно трудно проникают в мозг, заметно возрастает.
Распределение лекарственного средства в организме с учетом всех факторов, влияющих на этот процесс, характеризуется фармакокинетическим показателем — объемом распределения—Vd(Volumofdistribution). Это условный объем жидкости, необходимый для равномерного распределения в нем лекарственного средства, обнаруживаемого в терапевтической концентрации в плазме крови после однократного внутривенного введения, определяемый по формуле
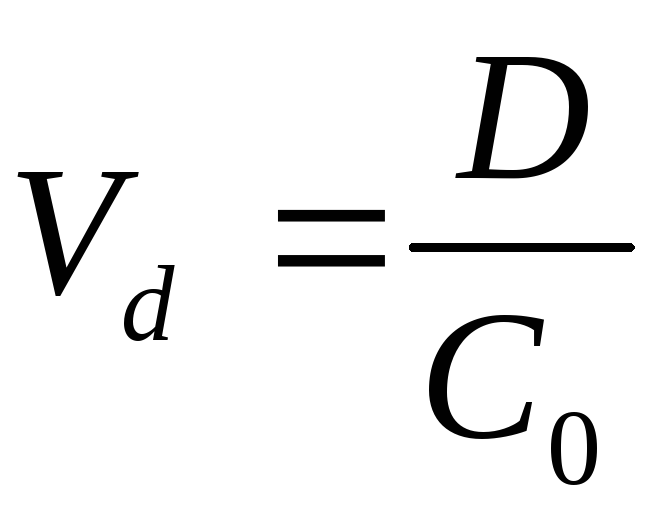 ;
;
где Vd— объем распределения;
D— введенная доза лекарственного вещества,
С0— начальная концентрация в крови.
Объем распределения дает представление о фракции вещества, находящейся в плазме крови. Для липофильных соединений, легко проникающих через тканевые барьеры и имеющих широкое распределение, характерно высокое значение Vd.. Если вещество в основном циркулирует в крови,Vdимеет низкие величины. Данный параметр важен для рационального дозирования веществ.
Если для условного человека с массой тела 70 кг Vd = 3 л (объем плазмы крови), это означает, что вещество находится в плазме крови, не проникает в форменные элементы крови и не выходит за пределы кровеносного русла.
Vd = 15 л означает, что вещество находится в плазме крови (3 л), в межклеточной жидкости (12 л) и не проникает в клетки тканей.
Vd = 40 л (общее количество жидкости в организме) означает, что вещество распределено во внеклеточной и внутриклеточной жидкости.
Vd = 400–600–1000 л означает, что вещество депонировано в периферических тканях и его концентрация в крови низкая. Например, для имипрамина (трициклический антидепрессант) Vd = 1600 л. В связи с этим концентрация имипрамина в крови очень низкая и при отравлении имипрамином гемодиализ не эффективен.
Источник
Распределение лекарственных веществ в организме
После попадания в системный кровоток лекарственное вещество распределяется по различным тканям организма. Характер распределения лекарственного средства определяется растворимостью его в липидах, степенью связывания с белками плазмы крови, интенсивностью регионарного кровотока и другими факторами. Большая часть лекарственного вещества в первые минуты после всасывания попадает в те органы и ткани, которые наиболее активно кровоснабжаются — сердце, печень, почки. Медленнее происходит насыщение лекарственным препаратом мышц, слизистых оболочек, кожи и жировой ткани. Для достижения терапевтических концентраций лекарственных веществ в этих тканях требуется от нескольких минут до нескольких часов. Важным фактором, определяющим распределение лекарственного вещества, является скорость его диффузии в различные ткани. Легко и быстро происходит диффузия в интерстициальную ткань. Капилляры хорошо проницаемы и для водорастворимых, и для жирорастворимых веществ, поэтому водорастворимые препараты (например, стрептомицин), которые плохо всасываются из кишечника, вводят парентерально. Они хорошо проникают во внеклеточные области, но не оказывают действия на ЦНС и другие органы, попасть в которые вещество может только преодолев мембранные барьеры. Растворимые в жирах препараты (например, газообразные анестетики) быстро распределяются по всему организму, одинаково хорошо проникая во внеклеточные и внутриклеточные области.
Связывание лекарственных веществ с белками крови и тканей
Многие лекарственные вещества обладают выраженным физико-химическим сродством к различным белкам плазмы крови, прежде всего к альбумину. Связывание лекарственных веществ с белками плазмы приводит к снижению их концентрации в тканях и месте действия, так как только свободный (несвязанный) препарат проходит через мембраны. Вещество, находящееся в комплексе с белком, лишено специфической активности. Свободная и связанная части лекарственного средства находятся в состоянии динамического равновесия. Иногда лекарственные вещества накапливаются в тканях в больших концентрациях, чем можно было бы ожидать, исходя из диффузионного равновесия. Этот эффект зависит от градиента рН, связывания лекарственного средства с внутриклеточными элементами и его распределения в жировой ткани. Клиническое значение имеют случаи, когда с белками крови связывается более 90% лекарственного вещества.
Нарушение связывания лекарственных веществ наблюдается при снижении концентрации альбуминов в крови (гипоальбуминемия) и связывающей способности белков крови при некоторых заболеваниях печени и почек. Даже снижение уровня альбуминов в крови до 30 г/л (в норме 33-55 г/л) может привести к значительному повышению содержания свободной фракции фенитоина. Клинически значимое увеличение уровня свободной фракции фуросемида происходит при снижении количества альбумина до 20 г/л.
Биотрансформация лекарственных средств Понятие и механизмы биотрансформации
Под биотрансформацией, или метаболизмом, понимают комплекс физико-химических и биохимических превращений лекарственных средств, в процессе которых образуются полярные водорастворимые вещества (метаболиты), которые легче выводятся из организма. В большинстве случаев метаболиты лекарственных средств менее биологически активны и менее токсичны, чем исходные соединения. Однако биотрансформация некоторых веществ приводит к образованию метаболитов, более активных по сравнению с введенными в организм веществами.
Различают два типа реакций метаболизма лекарственных препаратов в организме: несинтетические и синтетические. Несинтетические реакции метаболизма лекарственных препаратов можно разделить на две группы: катализируемые ферментами эндоплазматического ретикулума (микросомальные) и катализируемые ферментами другой локализации (немикросомальные). К несинтетическим реакциям относятся окисление, восстановление и гидролиз. В основе синтетических реакций лежит конъюнгация лекарственных средств с эндогенными субстратами (глюкуроновая кислота, сульфаты, глицин, глутатион, метильные группы и вода). Соединение этих веществ с лекарственными препаратами происходит через ряд функциональных групп: гидроксильную, карбоксильную, аминную, эпоксидную. После завершения реакции молекула препарата становится более полярной и, следовательно, легче выводится из организма.
Все лекарственные средства, вводимые внутрь, до поступления в системный кровоток проходят через печень, поэтому их разделяют на две группы — с высоким и с низким печеночным клиренсом. Для лекарственных веществ первой группы характерна высокая степень экстракции гепатоцитами из крови. Способность печени метаболизировать эти препараты зависит от скорости кровотока. Печеночный клиренс лекарственных веществ второй группы зависит не от скорости кровотока, а от емкости ферментативных систем печени, метаболизирующих данные препараты. Последние могут обладать высокой (дифенин, хинидин, толбутамид) или низкой степенью связывания с белками (теофиллин, парацетамол). Метаболизм веществ с низким печеночным клиренсом и высокой способностью к связыванию с белками зависит прежде всего от скорости их связывания с белками, а не от скорости кровотока в печени.
На биотрансформацию лекарственных средств в организме влияют возраст, пол, окружающая среда, характер питания, заболевания и т.д.
Печень является основным органом метаболизма лекарственных веществ, поэтому любое ее патологическое состояние отражается на фармакокинетике препаратов. При циррозах печени нарушается не только функция гепатоцитов, но и ее кровообращение. При этом особенно изменяется фармакокинетика и биодоступность препаратов с высоким печеночным клиренсом (табл. 6.1,6.2). Увеличение биодоступности лекарственных средств с высоким печеночным клиренсом при пероральном применении больными циррозом печени объясняется, с одной стороны, снижением метаболизма, с другой — наличием портокавальных анастомозов, по которым препарат поступает в системное кровообращение, минуя печень. Метаболизм препаратов с высоким печеночным клиренсом, введенных внутривенно, снижен у больных циррозом печени, однако степень такого снижения очень различна. Колебание этого параметра зависит скорее всего от способности гепатоцитов метаболизировать лекарственные средства в зависимости от характера кровотока в печени.
 Типы реакций метаболизма лекарственных средств
Типы реакций метаболизма лекарственных средств
Метаболизм веществ с низким печеночным клиренсом, таких как теофиллин и диазепам, также изменяется при циррозе. В тяжелых случаях, когда снижается концентрация альбумина в крови, перестраивается метаболизм кислых препаратов, активно связывающихся с белками (например, фенитоина и толбутамида), поскольку возрастает концентрация свободной фракции препаратов. В целом при заболеваниях печени клиренс лекарственных средств обычно уменьшается, а период их полувыведения возрастает в результате снижения кровотока в печени и экстракции их гепатоцитами, а также увеличения объема распределения препарата. В свою очередь, уменьшение экстракции лекарств гепатоцитами обусловлено снижением активности ферментов, нарушением захвата молекул лекарственных средств и/или связывания их с тканями печени и белками плазмы крови.
Принципы дозирования лекартсвенных веществ при заболеваниях печени
Необходимо помнить, что при поражении печени усиливается токсическое влияние многих лекарственных веществ на ЦНС и резко возрастает частота энцефалопатий. При заболеваниях печени (в зависимости от их тяжести) некоторые лекарственные средства противопоказаны либо их следует применять с осторожностью (барбитураты, наркотические анальгетики, ингибиторы моноаминоксидазы, фенотиазины, андрогенные стероиды и т.д.).
Источник


