- Производство новых лекарственных средств
- Как создать препарат в лаборатории и перенести его в производство
- Как с помощью ИТ появляются новые лекарства: мишени, ключи и миллиарды долларов
- Прицельное попадание в мишень
- ИТ и математика в фармацевтике
- Доклинические исследования
- Клинические испытания
- А сколько всё это стоит?
Производство новых лекарственных средств
Разработка лекарственного средства начинается с синтеза новых химических соединений. Вещества со сложной структурой можно получить из различных иа очников, например растений (сердечные гликозиды), тканей животных (гепарин), микробных культур (бензилпенициллин), культур человеческих клеток (урокиназа), или посредством генно-инженерных технологий (человеческий инсулин).
Чем больше ясности во взаимоотношениях структуры и активности, тем более направленным оказывается поиск новых веществ.
а) Доклинические испытания дают информацию о биологических эффектах новых веществ. Начальный скрининг может включать биохимические и фармакологические исследования (анализ связывания с рецептором) или эксперименты на культурах клеток, изолированных клетках и изолированных органах.
Поскольку эти модели не способны воспроизвести сложные биологические процессы, происходящие в интактных организмах, любое потенциальное лекарственное средство необходимо проверить на животных. Только эксперименты на животных позволяют выяснить, возникаютли желаемые эффекты при дозах, не вызывающих токсичности или сопровождающихся слабой токсичностью. Цель токсикологических исследований заключается в том, чтобы оценить:
1) токсичность, обусловленную кратковременным или длительным приемом;
2) генетические повреждения (генотоксичность, мутагенез);
3) развитие опухолей (канцерогенность);
4) возникновение врожденных дефектов (тератогенность).
В экспериментах на животных также оценивают всасывание, распределение, метаболизм и элиминацию (фармакокинетика) изучаемых веществ. На уровне доклинического изучение лишь у малой части новых веществ обнаруживается потенциал для применения у человека.
Фармацевтическиетехнологии предлагают методы изготовления лекарственных форм.
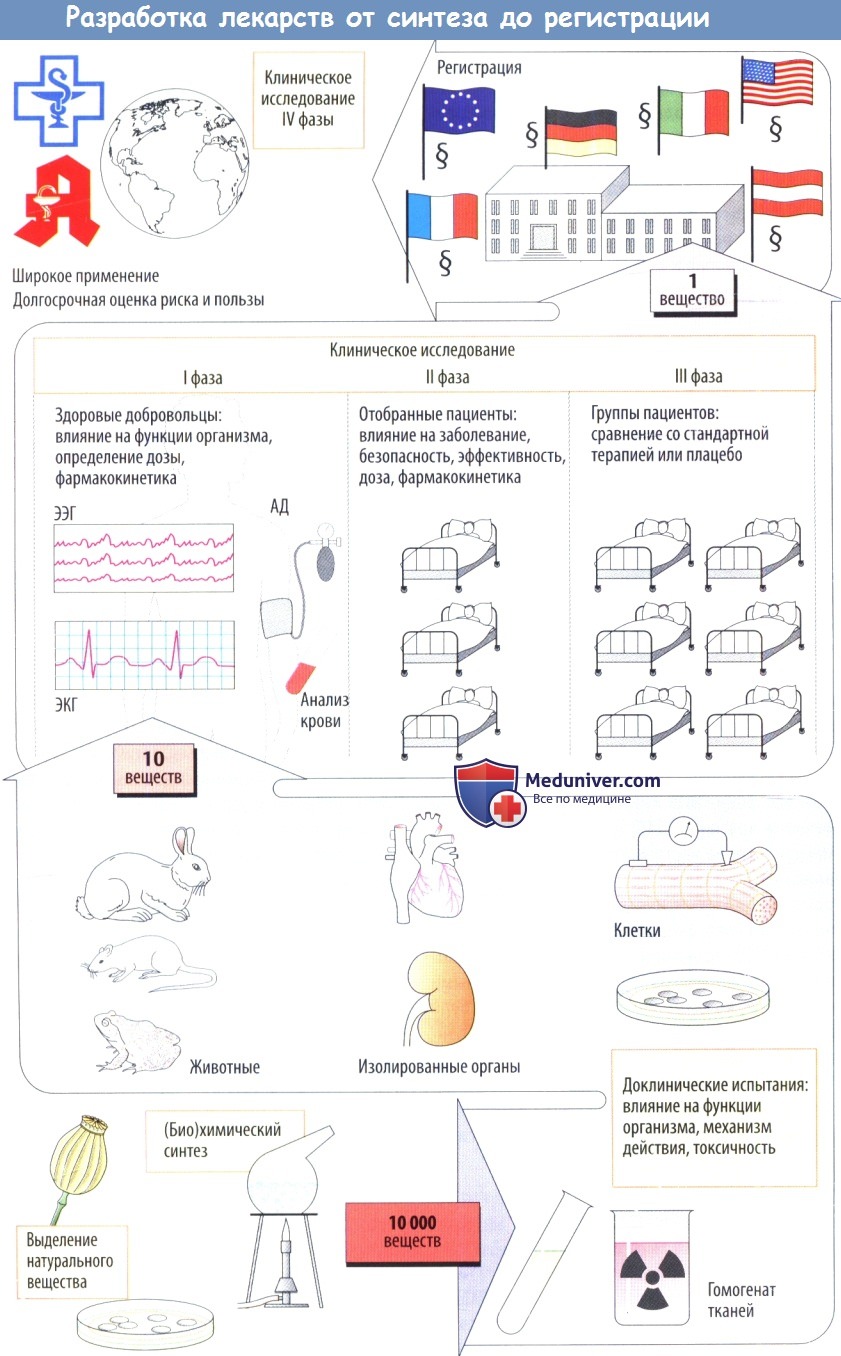
б) Клинические испытания начинаются с исследований I фазы, в которых участвуют здоровые лица; цель этих исследований — определить, будут ли эффекты, наблюдаемые у животных, также возникать у людей. Кроме того, на данном этапе определяется дозозависимость клинических эффектов.
Во II фазе потенциальные лекарственные средства сначала проверяют на отобранных пациентах на терапевтическую эффективность при заболевании, для лечения которого эти препараты предназначались. Если полезное действие очевидно, а частота побочных эффектов приемлема, начинается III фаза, в которой участвует более крупная группа пациентов, у которых новое средство сравнивают с традиционными методами лечения сточки зрения терапевтического результата.
Как форма экспериментов на людях, эти клинические исследования подвергаются анализу и одобрению этическими комитетами медицинских учреждений в соответствии с международными правилами проведения (Хельсинкской, Токийской и Венецианской декларациями). Во время клинических исследований выясняется, что многие вещества нельзя использовать. Как правило, в конце концов примерно из 10 000 вновь синтезированных веществ остается только одно.
в) Решение зарегистрировать новое лекарственное средство выносится национальным регуляторным органом (Food and Drug Administration в США, Health Protection Branch Drugs Directorate в Канаде, комиссией ЕС вместе с European Medicines Agency в Великобритании), которому производители должны подавать регистрационные документы.
Заявители должны документально подтвердить результатами соответствующих испытаний (доклинических и клинических), что критерии эффективности и безопасности удовлетворены и что лекарственные формы продукта (таблетки, капсулы и т. д.) соответствуют всем стандартам контроля качества.
После регистрации новое лекарственное средство может продаваться под торговым названием, оно должно быть доступным, выписываться врачами и отпускаться фармацевтами. В это время наблюдение продолжается в форме постмаркетинговых исследований (IV фаза клинических исследований)
г) Фармакологический надзор — действия, направленные на то, чтобы выявлять и устранять связанные с препаратом риски во время проведения клинических исследований и последующего его выхода на рынок. Фармаконадзор включает отчеты о предполагаемых случаях нежелательных реакций, направляемые в национальные регуляторные органы.
На основе длительного опыта применения можно правильно оценить соотношение риска и пользы и, следовательно, терапевтическую ценность нового лекарственного средства. Если новый препарат имеет небольшое преимущество перед существующими, необходимо принимать во внимание соотношение затрат и пользы от применения лекарственного средства.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Источник
Как создать препарат в лаборатории и перенести его в производство
Все мы ходим в аптеку и покупаем лекарства: таблетки, порошки, мази, растворы и многие другие формы лекарственных веществ. Но мало кто знает, как происходит создание лекарств и какой путь необходимо пройти от научной разработки в руках ученого до получения регистрационного досье на готовый препарат.
Разработка и создание лекарственных препаратов проходят при финансовой поддержке различных государственных и коммерческих структур (фондов) в соответствии с утвержденными приоритетными направлениями развития науки, технологий и техники в Российской Федерации, согласно перечню (указ президента России от 7 июля 2011 года №899). Одним из таких направлений являются «Технологии снижения потерь от социально значимых заболеваний».
Терапия и диагностика онкологических заболеваний — одно из приоритетных направлений. Многие ученые работают над созданием новых низкомолекулярных веществ для химиотерапии, получением новых аналогов уже существующих препаратов для преодоления возникающей резистентности опухолевых клеток, а также созданием новых лекарственных форм препаратов для улучшения биодоступности активного компонента и уменьшения побочных эффектов. В последние годы широко развивается направление адресной доставки препаратов — например, на основе антител.
Нашей научной группой под руководством члена-корреспондента РАН, профессора, доктора химических наук Евгения Северина разработан универсальный подход к созданию препарата для адресной доставки в злокачественные новообразования. Данный метод заключается в синтезе наночастиц, содержащих активное вещество, с последующей конъюгацией векторной молекулой — белка, способного связываться с рецепторами на поверхности опухолевых клеток. Предварительные исследования показали многообещающие результаты, проведенные доклинические испытания подтвердили, что разработанный прототип препарата обладает большей противоопухолевой активностью по сравнению с аналогом, представленным на рынке. Суммируя опыт проведения таких исследований, мы можем описать стандартный протокол проведения исследований при разработке нового препарата и оформления нормативной документации.
Каков же общий путь исследований, позволяющих провести доказательную базу эффективности и безопасности нового препарата, и какое количество времени для этого необходимо? После проведения этапов разработки подхода создания препарата и проведения предварительных экспериментов для доказательства его эффективности коллективом оформляется заявка для участия в конкурсе для предоставления финансирования на проведение доклинических испытаний. Стандартный грант предоставляется на три года, по результатам выполнения которого у группы исследователей, выполняющих данную работу, будет готовый прототип препарата, изучена его эффективность, безопасность и оформлены все необходимые нормативные документы, на основе которых формируется досье и подается на рассмотрение в Министерство здравоохранения России.
Первое, что необходимо сделать при разработке нового препарата,— проведение обширного литературного и патентного поиска в близких и смежных областях, чтобы избежать «изобретения велосипеда». Если патентная чистота подтверждена, можно приступать к экспериментальной работе.
В полученном гранте на проведение доклинических испытаний прописан календарный план и список этапов, которые необходимо выполнить, а затем подготовить отчетную документацию. Все этапы регламентированы соответствующими нормативными документами. Настольными книгами для специалистов являются «Государственная фармакопея Российской Федерации» и «Руководство по проведению доклинических исследований лекарственных средств» под редакцией А. Н. Миронова. В фармакопее прописаны все требования и нормы к разрабатываемым препаратам, какие виды исследования необходимо провести для подтверждения состава, структуры и свойств будущего лекарства или новой лекарственной формы (порошки, таблетки, растворы и пр.). В руководстве по проведению доклинических исследований подробно изложено, как необходимо проводить доклинические испытания, чтобы исследование было стандартизировано: выбор вида животных, их количества, кратность введения, дозы и пр.
Для проведения такого широкого спектра исследований необходимо соответствующее количество будущего препарата. В лабораторных условиях обычно отрабатывают технологические режимы и оптимальные параметры получения — от температурного режима до масштабирования процесса — и изучают влияние этих параметров на свойства получаемого продукта. По оптимизированным условиям пишут лабораторный регламент, где четко описано, как именно и при каких условиях необходимо проводить каждый этап получения препарата и что должно быть на выходе, вплоть до учета потерь производства. Лабораторный регламент является официальным нормативным документом, на его основе составляют опытно-промышленный и промышленный регламенты. Для последних двух необходима специальная производственная площадка, аттестованная под изготовление похожего продукта, как и разработанный препарат (рекомбинантные белки, вакцинные препараты, противоопухолевые препараты и т. д.). Необходимо перенести лабораторную технологию получения продукта в больший масштаб на промышленное оборудование, отработать регламентированный процесс получения и оптимизировать технологию с учетом новых объемов. Таким образом, на промышленных производственных площадках по опытно-промышленному и промышленному регламенту получают опытные партии препарата, которые затем необходимо проверять на соответствие всем показателям, которые были установлены разработчиками после получения оптимальной партии по лабораторному регламенту. Все требования к полученному препарату описаны в нормативном документе — фармацевтической статье предприятия (ФСП).
ФСП пишут по тем методам, с помощью которых анализируют полученный препарат в соответствии с государственной фармакопеей. Для включения метода в ФСП необходимо использовать либо стандартизированные методы, приведенные в фармакопее, либо валидированные методы, которые были использованы, но отсутствуют в фармакопее или отличаются по условиям проведения от описанных. Чем больше активных (целевых) и вспомогательных компонентов в препарате, тем больше методов содержит ФСП. Необходимо провести количественный анализ активного компонента и примесей, полный качественный анализ, а также подтвердить сохранение функциональной активности основного компонента.
Зачем нужна фармацевтическая статья предприятия? В технологическом процессе получения препарата могут возникнуть непредвиденные неполадки на какой-либо стадии производства. Для выявления несоответствия продукта (брака производства) необходимо проводить анализ каждой партии. Если была получена бракованная партия, ее можно будет легко выявить, проведя все анализы и сравнив с установленными нормами в ФСП.
После получения опытных партий необходимо изучить стабильность полученного препарата, чтобы доказать, что за указанный промежуток времени (срок годности) не происходит никаких существенных изменений и свойства будущего лекарства остаются неизменными. Стандартный срок хранения лекарственных средств — от полугода до трех лет. Для противоопухолевых препаратов — два года. Для конкурентоспособности и рентабельности будущего лекарства необходимо придерживаться таких же сроков годности, а также ориентироваться на аналоги. Однако ждать два года, чтобы узнать, стабилен ли препарат и проходит ли он по всем нормам и стандартам, нет необходимости. Существуют протоколы, описанные в фармакопее (ОФС.1.1.0009.15 «Сроки годности лекарственных средств»), позволяющие сократить период исследования до года и даже шести месяцев, используя более агрессивные условия исследования. Если препарат по всем показателям сохраняет количественные, качественные и функциональные характеристики, указанные в ФСП, следующий этап — проведение доклинических исследований на животных. На все перечисленные стадии оформляют нормативные документы: регламенты на производство, отчеты о валидации, акты наработки, ФСП, протоколы анализа партий, подтверждающие соответствие полученного продукта описанному в документах.
Для изучения безопасности и эффективности полученного препарата на лабораторных животных утверждается план доклинических исследований, в котором перечислены все этапы исследования и их последовательность. План и модели проведения исследований составляются в зависимости от типа разработанного препарата и описаны в «Руководстве по проведению доклинических исследований лекарственных средств». Для оформления регистрационного досье необходимо провести полные доклинические исследования: исследование нескольких видов токсичности (общетоксическое действие, аллергизирующие свойства, иммунотоксическое действие, репродуктивная токсичность и др.), эффективности действия (например, противоопухолевый эффект нового препарата в сравнении с аналогами), изучить фармакокинетику. Все полученные данные обрабатывают статистически и оформляют в нормативный документ — отчет о доклинических исследованиях с прикреплением первичных результатов. На основании полученных данных, в случае если препарат обладает эффективностью и при этом безопасен для применения, разработчики пишут план проведения первой стадии клинических испытаний, проект инструкции по применению и проект брошюры исследователя. Из составленных документов формируется регистрационное досье, которое и подается на рассмотрение в Министерство здравоохранения России с другими сопутствующими документами.
До последнего этапа, описанного в данной статье, доходят немногие разработки. Путь от научной идеи до регистрации может занимать от трех лет до десятилетий. При наличии оформившейся идеи, прошедшей предварительные фундаментальные исследования, все описанные этапы исследования можно провести за три-пять лет. А дальше препарат ждет еще более сложный, но не менее интересный путь: клинические испытания, регистрация, производство и выход на рынок — при условии наличия хороших результатов на этапе клинических испытаний.
Елена Никольская, кандидат химических наук, старший научный сотрудник ИБХФ РАН
Источник
Как с помощью ИТ появляются новые лекарства: мишени, ключи и миллиарды долларов
Фармацевтическая индустрия бьет рекорды — с каждым годом выпускается всё больше новых лекарственных препаратов. В 2018 году появилось 68 новых активных фармацевтических субстанций (АФС) — веществ или их смесей, предназначенных для использования в производстве лекарственного препарата. За 9 месяцев 2019 года импорт фармсубстанций в РФ превысил объем поставок за весь 2018 год. Глава евразийского подразделения фармацевтической компании «Босналек» Валентина Бучнева рассказала об этапах разработки лекарств и о том, как появляются новые АФС.
Читайте «Хайтек» в
Разработка новых лекарственных препаратов — долгий и сложный процесс. От исследований в лаборатории до попадания в аптеки и больницы проходит несколько лет — в среднем от 10 до 14. Срок в пять лет — удача для производителя, случается такое крайне редко. Плюс ко всему, из нескольких тысяч веществ, которые исследуются фармацевтами на первых этапах, до аптеки доходит всего одно.
Над созданием препаратов работает команда из нескольких сотен человек. Это биологи, генетики, химики и медики. Первый этап работы — лабораторные исследования.
Прицельное попадание в мишень
Перед тем, как начать разрабатывать лекарство от какого-либо недуга, нужно точно знать причину болезни и ключевую точку, при воздействии на которую болезнь можно остановить и повернуть процесс вспять. Эта точка называется мишенью. Чаще всего это белок или какой-то фермент в организме человека или в болезнетворном организме.
Когда мишень выбрана, начинается формирование целевой терапевтической молекулы — прообраза. Он должен соответствовать поставленным целям и успешно попадать в мишень.
Обычно подходит лишь одно действующее вещество. Для того, чтобы его найти, необходимо перебрать тысячи или даже десятки тысяч вариантов. Сейчас этот процесс выполняется при помощи компьютерных технологий, раньше фармацевты действовали фактически вслепую, методом проб и ошибок, на что уходило гораздо больше времени.
ИТ и математика в фармацевтике
Цифровые технологии позволяют значительно ускорить процесс разработки лекарственных препаратов. Особенно эффективно использование ИТ на начальных стадиях работы по созданию препарата. Для некоторых видов фармацевтических препаратов их лечебные свойства можно предсказать еще на стадии моделирования. Дело в том, что ученым известны параметры модели — физиологические процессы заболевания и мишень. Моделирование дает возможность собрать «ключ» — молекулу, которая идеально подходит для мишени.
Выгода от использования компьютерных технологий еще и в том, что они помогают специалистам обрабатывать огромные массивы данных. Фармацевтические компании обладают гигантскими базами, в которых содержится подробная информация о разработке и свойствах разных препаратов. Мозг человека просто не в состоянии охватить всё это, поэтому на помощь пришли математика, компьютерный анализ, big data и машинное обучение.
В частности, математическая модель ускорила выход на рынок препарата против рассеянного склероза. Его разработчики определили точную концентрацию активного вещества для оказания максимального положительного эффекта на больного. Регулятор из США, FDA, согласился с выкладками ученых и одобрил регистрацию препарата без дополнительных исследований. Пока что это исключение из общего правила, но его можно назвать прецедентным.
Доклинические исследования
Следующий этап — поиск наиболее эффективного действующего вещества. Оптимальный вариант выбирается в ходе тестирования нескольких активных веществ на животных. Фаза называется доклиническими исследованиями. Специалисты оценивают силу биологического действия вещества, наблюдают за возможными побочными эффектами. Это нужно для того, чтобы определить показания к применению препаратов и идентифицировать противопоказания.
Кроме того, доклинический этап исследования позволяет исследователям определить дозозависимость эффектов лекарства плюс максимально возможную безопасную дозу и целесообразность ее повышения. Если доза будет очень большой, лекарство станет токсичным, слишком маленькой — оно может не оказать необходимого лечебного эффекта. Задача — определить терапевтическое окно, то есть такой диапазон доз, когда лекарство дает максимальный эффект, но еще не является токсичным.
На этом этапе тоже используются компьютерные технологии, что снижает количество подопытных животных. К сожалению, полностью отказаться от таких испытаний фармацевты пока не могут. Исследование новых препаратов включает в себя определение общей и специфической токсичности, фармакодинамики (то, как ведет себя препарат в организме) и фармакокинетики (то, как влияет препарат на организм в целом). Математические методы позволяют построить модель как фармакокинетики, так и фармакодинамики. Модель обычно представляет сложную систему, которая учитывает множество факторов.
В ходе доклинических испытаний может отсеяться около 90% потенциально эффективных препаратов.
Клинические испытания
После того, как разработчики препарата провели доклинические испытания, выяснив все важные моменты, наступает стадия клинических испытаний. В ее ходе может отсеяться еще 90% действующих веществ.
Структура клинических испытаний зависит от вида препарата. Обычно выделяют четыре основных этапа клинических испытаний.
- Тестирование препарата на здоровых добровольцах. Как правило, их не должно быть меньше 10. Добровольцы нужны для того, чтобы точно выяснить побочные эффекты, определить переносимость и безопасность препарата. Специалисты на этом этапе проводят новое изучение фармакокинетики и фармакодинамики, но уже на организме человека, а не животных.
- Проверка лечебного воздействия препаратов на пациентах с конкретным заболеванием. Чаще всего добровольцев разделяют на основную группу, которая принимает лекарства, и контрольную группу, которой дают плацебо — лекарство без действующего вещества.
- Масштабная проверка препарата на больших группах добровольцев. Численность таких групп может составлять тысячу человек и больше. Испытуемые должны быть разного возраста, с сопутствующими заболеваниями .
Если всё хорошо, подается заявка на регистрацию препарата (это тысячи документов, отчеты исследователей, результаты доклинических и клинических испытаний). Если у регистрирующих органов возникают какие-то сомнения, заявку могут отправить на доработку или вообще запретить вывод препарата на рынок.
- Пострегистрационный этап, он проводится уже после того, как лекарство выпустили на рынок. Цель — определить эффективность препарата по отношению к аналогам (если они есть), выявить дополнительные побочные эффекты и факторы риска. Если будут найдены серьезные проблемы, лекарство может быть даже отозвано с рынка.
А сколько всё это стоит?
Очень много. По оценке аналитиков, средние затраты фармацевтических компаний на вывод нового лекарственного препарата на рынок составили $802 млн в 2000 году. Через несколько лет сумма увеличилась до $1,7 млрд, сейчас этот показатель еще выше и составляет около $2,5 млрд. А вот вероятность появления на рынке новой молекулы после клинических испытаний составляет всего 11,5%. То есть девять лекарственных препаратов из десяти никогда не попадут в аптеки, несмотря на то, что компании вложили в разработку огромные деньги.
Стоимость исследований постоянно увеличивается, а вот их результативность — падает . По этой причине фармацевтические компании адаптируют свои бизнес-модели к текущим условиям, учитывая такие факторы, как небольшая коммерческая отдача от стадии исследований, замедление роста выручки и конкуренция со стороны других компаний и дженериков (аналоги оригинальных лекарственных препаратов).
Проблема высокой стоимости этапа исследований решается при помощи концепции открытых инноваций — это вовлечение партнеров со стороны в процесс исследований и разработок. Фармацевтические компании активно сотрудничают с научным сообществом, стартапами и институтами развития. В итоге компании помогают исследователям в их разработках, а ученые — приносят пользу фармацевтике за счет новых идей, методов работы, оригинальных решений.
Что касается западных стран, то концепция открытых инноваций там активно работает. В России же она находится на этапе становления. Отечественные фармкомпании начинают активно работать с учеными, научно-исследовательскими организациями и государством. Если всё получится, то российским ученым удастся создать эффективную цепочку производства лекарственных препаратов, ускорив появление новых лекарств и снизив стоимость всего цикла разработки.
Источник


