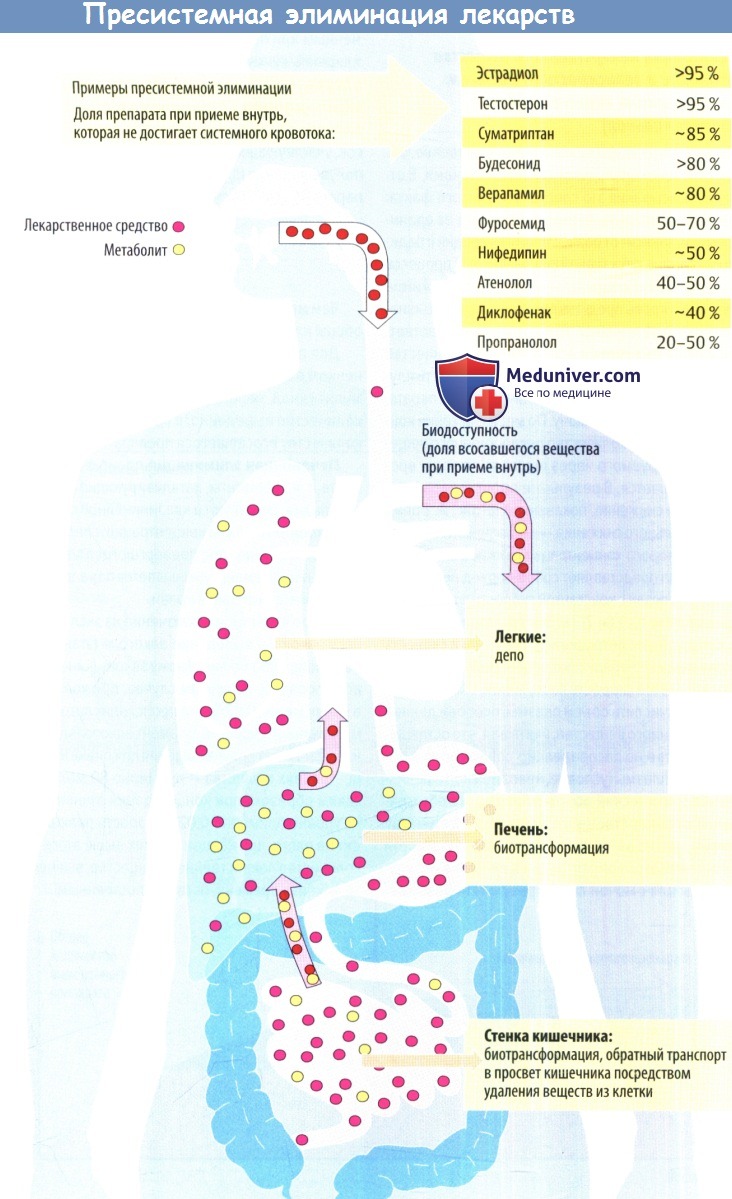
- Пресистемная элиминация лекарственных веществ это
- Пресистемная элиминация
- 1.Всасывание лекарств. Пресистемная элиминация. Пролекарства. Сравнительная биодоступность при сублингвальном, оральном и ректальном способах введения.
- Связь «доза-эффект»
- Выбор пути введения лекарств
- Связывание с белками плазмы крови и распределение лекарственных средств
- Элиминация лекарственных средств
Пресистемная элиминация лекарственных веществ это
Морфологические барьеры организма проиллюстрированы в предыдущей статье на сайте. Физико-химические свойства лекарственного средства определяют, достигнет ли он цели, расположенной на поверхности или внутри клеток организма либо бактериальных клеток, и в какой степени.
В тех случаях, когда препарат принимается внутрь или вводится парентерально, фармакокинетические процессы идут совершенно иначе, чем при местном применении.
Это становится очевидным, если проследить путь принятого внутрь препарата, начинаяс места всасывания до попадания в кровоток. Возможен один из следующих вариантов развития событий.
1. Препарат проникает через эпителий кишечника в энтероциты, однако Р-гликопротеид транспортирует его обратно в просвет кишечника. Поэтому итоговое всосавшееся количество препарата может быть значительно меньше.
Этот «противотранспорт» различен у разных людей в отношении одного и того же вещества и, кроме того, подвержен влиянию других лекарственных средств.
2. На пути из просвета кишечника в общее циркуляторное русло принятый внутрь препарат расщепляется под воздействием ферментов, например цитохромоксидазы Р450.
(а) Разрушение может начаться уже в слизистой кишечника. Другие лекарственные средства или химические вещества могут подавлять или стимулировать активность изоферментов цитохрома в кишечнике.
Отдельный пример — сок грейпфрута, который подавляет активность оксидаз CYP3A4 в стенке кишечника и вызывает повышение в крови концентрации некоторых лекарственных средств до токсического уровня.
(б) Самую важную роль играет метаболизм в печени, которому подвергается любое лекарственное средство, попадающее в организм. В печени работает множество ферментов, химически преобразующих эндогенные и экзогенные вещества, чтобы обеспечить их выведение из организма. Только часть всосавшегося количества может попасть в кровь печеночной вены, это зависит от количества препарата, поглощенного и переработанного гепатоцитами.
Следует учитывать, что другие препараты могут вызывать повышение активности ферментов (увеличение гЭР).
Процессы, о которых говорится выше, объединены термином «пресистемная элиминация».
3. Парентеральное введение лекарственного вещества позволяет обойти пресистемную элиминацию. При в/в, п/к и в/м введении препарат проходит через полую вену, попадает в правый желудочек и, через легкие, в левый желудочек, далее — в системный кровоток.
Богатые жиром и имеющие большую поверхность легкие всасывают некоторое количество липофильных и амфифильных веществ, а затем медленно выделяют их после снижения концентрации в крови.
Во время быстрого поступления лекарственного средства в организм легкие играют роль буфера и защищают сердце от избыточной концентрации веществ после в/в введения.
В некоторых ситуациях желательна быстрая пресистемная элиминация. Яркий пример — назначение глюкокортикоидов при бронхиальной астме.
Поскольку большая часть ингалированного препарата обычно проглатывается, глюкокортикоиды с полной пресистемной элиминацией оказывают минимальную системную нагрузку на организм.
Использование клопидогрела для ингибирования агрегации тромбоцитов — пример желательной пресистемной активации.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Источник
Пресистемная элиминация
При приеме препарата внутрь, кроме биологических мембран, стоящих на пути из просвета кишечника в кровь, имеется еще один фактор, ограничивающий поступление лекарства в системный кровоток – печеночный метаболизм. Лекарственное вещество, поступая из ЖКТ по воротной вене в печень, может подвергнуться ферментативному разрушению, в связи с чем в системный кровоток попадает лишь часть (иногда незначительная часть) принятой дозы. Данный феномен носит название«эффект первого прохождения через печень». Так, некоторые лекарственные средства, обладая высокой абсорбцией, попадают в системный кровоток в очень небольшом количестве, не оказывающем терапевтического эффекта. Этот эффект характерен для быстро метаболизирующихся (см. фазу биотрансформации) средств и при значительной выраженности исключает возможность приема соответствующего препарата внутрь (например, антиаритмик лидокаин). В других случаях его можно корригировать увеличением дозы, которая оказывается значительно выше, чем при внутривенном введении (верапамил, морфин, пропранолол). Весьма демонстративным в этом плане является пример нитроглицерина. Эффект первого прохождения через печень у этого препарата достигает 85-97% дозы, что объясняет необходимость его назначения «в обход» печени (сублингвально или внутривенно) и делает бессмысленным нередко встречающееся назначение внутрь «капель Вотчала» (раствор нитроглицерина в ментоле).
Часть введенной дозы, достигшая системного кровотока, является важнейшей характеристикой препарата. Последняя обозначается как «биодоступность» и по определению ВОЗ понимается как степень и скорость, с которой вещество или его активная часть доставляется из лекарственной формы в системный кровоток. При внутривенном введении биодоступность принимается за 100%. При приеме внутрь она зависит от ряда факторов: устойчивости лекарства к действию соляной кислоты желудочного сока, активности разрушения препарата ферментами в просвете и стенке кишечника, выраженности эффекта первого прохождения через печень, то есть от потерь вследствие, так называемой,пресистемной элиминации.
Пресистемная элиминация зависит не только от препарата, но и от ряда факторов организма пациента и условий применения лекарства. На пресистемную элиминацию влияют взаимодействия с пищей и другими лекарственными средствами (см. гл. 9), скорость эвакуации из желудка, моторная функция кишечника, состояние функции печени и портального кровообращения. При нарушении функции печени (при циррозе), а также при развитии системы анастомозов между воротной веной и полыми венами (при портальной гипертензии) лекарственное средство попадает в системный кровоток, минуя печень. При этом снижается эффект первого прохождения через печень, что может вести к передозировке лекарства несмотря на назначение терапевтической дозы.
Кроме перечисленных факторов на биодоступность влияют еще и особенности технологии приготовления лекарственной формы. Поэтому биодоступность одного и того же активного вещества, выпускаемого в разных лекарственных формах или в лекарствах различных производителей, может колебаться в широких пределах. Однако такие колебания существенно затрудняют эффективное и безопасное дозирование лекарственных средств. В связи с этим перед регистрацией лекарства необходимо провести исследование биодоступности нового препарата в сравнении с эталонным лекарственным средством. В результате получается информация о сравнительной биодоступности или о биоэквивалентности.
Тут вы можете оставить комментарий к выбранному абзацу или сообщить об ошибке.
Источник
1.Всасывание лекарств. Пресистемная элиминация. Пролекарства. Сравнительная биодоступность при сублингвальном, оральном и ректальном способах введения.
Пути введения ЛС: а. энтеральный путь введения: перорально, сублингвально, трансбуккально, ректально, через зонд; б. парентеральный путь введения: внутривенно, подкожно, внутримышечно, ингаляционно, субарахноидально, трансдермально.
Основные механизмы всасывания:
1.Пассивная диффузия через мембрану клеток. Определяется градиентом концентрации веществ.Чем выще липофильность вещества, тем легче оно всасывается через клеточную мембрану.
2. фильтрация через поры мембран. Диаметр пор в кишечнике не велик , поэтому через них диффундирует вода, некоторые оины, мелкие гидорофильные молекулы.
3 Активный транспорт характеризуется избирательностью к определенным соединениям, возможна конкуренция за один транспортный механизм, насыщаемостью. Активный транспорт обеспечивет всасывание гидрофильныхтполярных молекул.
4.При пиницитозе происходит инвагинация клеточной мембраны с последующим образованием пузырька( вакусли).Пузырек мигрирует по цитоплазме к противоположной стороне клетки, где путем экзоцитоза содержимое пузырька выводится наружу.
Пресистемная элиминация лекарственных средств (эффект первого прохождения) — процесс биотрансформации лекарства до попадания лекарственых средств в системный кровоток. В пресистемной элиминации при пероральном введении лекарства участвуют ферментативные системы кишечника, крови воротной вены и гепатоциты.
Сравнительная биодоступность: сублингвально- всасывание быстрое, минуя при первом пассаже печеночный барьер и не контактируя с ферментами ЖКТ. Орально- всасывание происходит частично из желудка, главным образом в тонкой кишке. Ректально- около 50% веществ поступает в кровоток, минуя печень. Всасывание – простой диффузии.
2 Нестероидные противовоспалительные средства: характеристика группы. К ним относятся вещества, оказывающие влияние на циклооксигеназу и таким путем снижают биосинтез простаноидов. ЦОК- продуцируется в обычных условиях и регулирует образование в организме простаноидов. ЦОК-2 в значительной степени индуцируется процессом воспаления( образуется в отсутствии воспаления).
I.неизбиратеные ингибиторы ЦОК -1 и -2:
Производные салициловой кислоты( кислота ацетилсалициловая)
Производные антраниловой кислоты( кислота мефенамовая, кислота флуфенамовая)
Производные индолуксусной кислоты( индометацин)
Производные фенилуксусной кислоты( диклофенак- натрий)
Производные фенилпропионовой кислоты(ибупрофен)
Производные нафтилпропионовой кислоты( напроксен)
Оксикамы( пироксикам, лорноксикам)
II.Избирательные ингибитры ЦОК-2 ( целекоксиб, рофекоксиб).
Эффект: Жаропонижающий эффект связан с уменьшением продукции простагландина Е2.нпвс действуют только при лихорадке
Анальгезирующий эффект связан с нарушением образования Е2 и I2,которые повышают чувствительность болевых рецепторов к брадикинину.
Фармакокинетика. Все НПВС хорошо всасываются в желудочно-кишечном тракте. Практически полностью связываются с альбуминами плазмы, вытесняя при этом некоторые другие лекарственные средства. Метаболизируются НПВС в печени, выделяются через почки.
Показания. Ревматические заболевания. Ревматизм (ревматическая лихорадка), ревматоидный артрит, подагрический и псориатический артриты, анкилозирующий спондилит (болезнь Бехтерева), синдром Рейтера.
Неревматические заболевания опорно-двигательного аппарата. Остеоартроз, миозит, тендовагинит, травма (бытовая, спортивная). Нередко при этих состояниях эффективно применение местных лекарственных форм НПВС (мази, кремы, гели).
Неврологические заболевания. Невралгия, радикулит, ишиас, люмбаго
Лихорадка (как правило, при температуре тела выше 38,5°С)..
противопоказаны при эрозивно-язвенных поражениях желудочно-кишечного тракта, особенно в стадии обострения, выраженных нарушениях функции печени и почек, цитопениях, индивидуальной непереносимости, беременности.
Побочные эффекты:из-за угнетения ПГ Е2 и I2 нарушается целостность слиз оболочки жедудка и 12-ти перстной кишки.тошнота.могут появится эрозии желудка.чтобы избежать это назначают нпвс совместно с препаратами гастропротекторных ПГ.
3 НадропаринПилокарпин: полная характеристика препарата.М-холиностимулирующее средство, оказывает миотическое и противоглаукомное действие.
Вызывает сокращение циркулярной (миоз) и цилиарной мышц (спазм аккомодации), увеличивает угол передней камеры глаза (оттягивается корень радужки), повышает проницаемость трабекулярной зоны (трабекула натягивается, и происходит открытие блокированных участков шлеммова канала), улучшает отток водянистой влаги из глаза и в конечном итоге снижает внутриглазное давление
Острый приступ закрытоугольной глаукомы, вторичная глаукома, первичная открытоугольная глаукома, абсцесс роговицы; кровоизлияние в стекловидное тело. Необходимость сужения зрачка после инстилляции мидриатиков.
Гиперчувствительность, ирит, циклит, иридоциклит, кератит, состояние после офтальмологических операций и др. заболевания глаз, при которых сужения зрачка нежелательно.
Источник
Связь «доза-эффект»
Является важным фармакодинамическим показателем. Обычно этот показатель представляет собой не простое арифметическое отношение и может графически выражаться по-разному: линейно, изогнутой вверх либо вниз кривой, сигмоидальной линией.
Каждое лекарство обладает рядом желательных и нежелательных свойств. Чаще всего при увеличении дозы лекарства до определенного предела желаемый эффект возрастает, но при этом могут возникать нежелательные эффекты. Лекарство может иметь не одну, а несколько кривых отношения «доза-эффект» для его различных сторон действия. Отношение доз лекарства, при которых вызывается нежелательный или желаемый эффект, используют для характеристики границы безопасности или терапевтического индекса препарата. Терапевтический индекс препарата можно рассчитывать по соотношению его концентраций в плазме крови, вызывающих нежелательные (побочные) эффекты, и концентраций, оказывающих терапевтическое действие, что более точно может характеризовать соотношение эффективности и риска применения данного лекарства.
Методы для изучения фармакодинамики
Методы для изучения фармакодинамики должны обладать рядом важных свойств:
а) высокой чувствительностью— способностью выявлять большую часть тех отклонений от исходного состояния, на которое пытаются воздействовать, а также оценивать положительные изменения в организме.
б) высокой специфичностью— способностью относительно редко давать «ложноположительные» результаты.
в) высокой воспроизводимостью— способностью данным методом стабильно отображать характеристики состояния больных при повторных исследованиях в одинаковых условиях у одних и тех же больных при отсутствии какой-либо динамики в состоянии этих больных по другим клиническим данным.
Фармакокинетика изучает особенности поступления препарата в организм в зависимости от пути введения, всасывания, связи с белками, плазмы крови, распределение и элиминацию лекарств и их метаболитов из организма.
Выбор пути введения лекарств
Выбор пути введения лекарств зависит от способности растворяться в воде или липидах, их действующего вещества, локализации патологического процесса и степени тяжести заболевания. По классификации академика АМН СССР В.М. Карасика все пути введения лекарственных средств можно разделить на 2 вида:
а ) без нарушения целостности кожных покровов — через рот (внутрь), через прямую кишку, ингаляционно, интраназально, трансдермально и т.п.;
б ) с нарушением целостности кожных покровов — подкожно, внутримышечно, внутривенно, в полости плевры, брюшины, суставов, интралюмбально, в желудочки мозга и т.п.
Лекарственные препараты могут преодолевать тканевые барьеры с помощью следующих механизмов:
1. Пассивная диффузия через «водные поры» по градиенту концентрации между эндотелиальными клетками капилляров только для солюбилизированных молекул, имеющих массу не более 30 000 дальтон. Между клетками эпидермиса, эпителия слизистой оболочки желудочно-кишечного тракта и т.п. промежутки меньше, и через них могут фильтроваться молекулы с массой не более 150 дальтон (например, ионы).
2.Пассивная диффузия через мембраны клеток по градиенту концентрации для липидорастворимых веществ. Это – наиболее важный механизм, так как для большинства лекарств характерна значительно большая растворимость в липидах, чем в воде.
Липидорастворимость препарата зависит от величины заряда его молекулы. Чем больше заряд, тем хуже вещество растворяется в жирах, и наоборот. Степень ионизации ксенобиотика зависит от рН среды, в которой он находится. Если препарат является слабой кислотой, то в кислой среде он будет находиться главным образом в неионизированном виде и лучше проникать через биологические мембраны, поэтому его надо назначать внутрь после еды, когда содержимое желудка максимально кислое.
И наоборот, лекарство, являющееся слабым основанием, правильнее назначать внутрь до еды (за 1-1,5 ч) или спустя 1,5-2 ч после еды, когда кислотность содержимого желудка минимальна. Важно учитывать наличие у больных нарушений кислотности (гипер — или гипоацидные состояния), а также возрастные особенности.
Например, рН в желудке на высоте секреции соляной кислоты составляете детей месячного возраста 5,8; в возрасте 3-7’мес около 5; 8-9 мес — 4,5; к 3 годам — 1,5-2,5, как у взрослых. Содержимое кишечника имеет слабощелочную реакцию (7,3-7,6).
Кроме того, лекарства — слабые кислоты лучше запивать кислыми растворами, а слабые основания — щелочными минеральными водами или молоком, которые к тому же ускоряют опорожнение желудка и поступление его содержимого в двенадцатиперстную кишку.
В плазме крови в физиологических условиях поддерживается рН 7,3-7,4. Однако при назначении лекарственных средств необходимо знать, что рН в некоторых жидких средах и тканях человека отличаются. Например, рН женского молока 6,4-6,7; слюны — 5,4-6,7; мочи — 4,8 (утром) — 7,4 (вечером) у старших детей и взрослых; клеток скелетных мышц 6,7-6,8; на поверхности кожи — 5,5; в очагах воспаления и некроза — кислая среда. При назначении препаратов это очень важно учитывать. Так, лекарство — слабое основание, попав в женское молоко, диссоциирует, что препятствует его возврату в кровь, и происходит его кумуляция в молоке, что представляет опасность при кормлении ребенка грудью. Лекарственное средство — слабая кислота, попав в мочу, имеющую кислую реакцию (утром), будет лучше реабсорбироваться, что, с одной стороны, может способствовать его задержке в организме, а с другой — уменьшать время нахождения препарата в моче, что нежелательно, если речь идет об использовании противомикробного препарата при инфекции мочевыделительной
2. Облегченная диффузия через мембраны клеток с помощью специальных носителей: белков-ферментов или транспортных белков. Так осуществляется перенос глюкозы в ткани или транспортом аминокислот через гематоэнцефалический барьер и плаценту.
3. Активный транспорт через клеточные мембраны против градиента концентрации с участием транспортных систем и с затратой энергии. У детей и людей пожилого возраста такой путь проникновения лекарств плохо развит. Работа данного активного механизма зависит от состояния сердечно-сосудистой системы, гемодинамики в конкретном органе или ткани.
4. Пиноцитоз — поглощение внеклеточного материала мембранами с образованием везикул. Этот процесс особенно важен для лекарственных средств полипептидной структуры с молекулярной массой более 1000 килодальтон.
Характеристика наиболее часто применяемых путей введения лекарств
Наиболее частым, удобным и, как правило, экономически выгодным путем введения лекарств в организм является их прием через рот (внутрь).
При приеме внутрь ксенобиотик, всасываясь, попадает в систему воротной (портальной) вены и в печень. Уже при первом прохождении через нее он может подвергнуться биотрансформации (подробнее — см. ниже). После этого та доля дозы лекарства от введенного внутрь его количества, которая поступает по полой вене в системный кровоток в активной форме, соответствует понятию биоусвояемость (или биодоступность) лекарства. Следует подчеркнуть, что инактивация препарата может происходить и в просвете желудочно-кишечного тракта под влиянием пищеварительных соков, которых за сутки вырабатывается 2-2,5 л; ферментов микрофлоры; некоторые лекарства могут связываться компонентами пищи. Биотрансформация ксенобиотика может происходить не только в печени, но и в других органах, в частности, в клетках слизистой оболочки желудочно-кишечного тракта. Весь же комплекс процессов, приводящих к инактивизации лекарственного вещества до его попадания в системный кровоток, называется пресистемной элиминацией.
При сублингвальном и суббукальном введении разновидности приема через рот препарат не подвергается воздействиям пищеварительных и микробных ферментов, быстро всасывается (эффект наступает в 2-3 раза быстрее, чем при приеме внутрь) и попадает в системный кровоток через верхнюю полую вену, минуя печень. Пресистемная элиминация при таком введении либо совсем отсутствует, либо очень мала.
К энтеральному пути введения лекарства также относится его назначение через прямую кишку — ректально. В прямой кишке нет пищеварительных ферментов, препарат после всасывания попадает в систему нижней полой вены и далее, минуя печень, в системный кровоток. Но существуют и отрицательные стороны этого способа введения: неудобство применения (особенно вне стационара); небольшая площадь всасывающей поверхности и порой непродолжительное время контакта лекарственного средства со слизистой оболочкой (ребенку бывает трудно удержать препарат в кишке); раздражающее действие (иногда возникает проктит).
В случае назначения пролекарства — неактивного вещества, которое должно под влиянием ферментных систем печени превратиться в активный препарат, его вводят только внутрь. В некоторых случаях возможна активация пролекарства ферментными системами крови, почек и т. п. В этих случаях возможны и другие пути введения.
Ингаляционно вводят газообразные вещества, жидкости и аэрозоли. При назначении последних очень важен размер твердых частичек. Частицы размером 60 мкм и больше оседают на поверхности глотки и заглатываются в желудок, размером 20 мкм проникают в терминальные бронхиолы, размером 6 мкм — в респираторные бронхиолы, размером 2 мкм — в предальвеолярный проход и 1 мкм — в альвеолы. Всасывание происходит в основном доза препарата, как правило, в несколько раз меньше, чем при приеме внутрь; быстрое наступления эффекта.
Имеются и некоторые недостатки инъекций:
— больше опасность передозировки (особенно при введении препаратов с малой широтой терапевтического действия);
—существует опасность возникновения тромбоза и гиперволемии (при внутривенном введении);
— в случае нарушения нормального локального кровотока или при токсикозах, обезвоживании, шоке, заболеваниях сердечно-сосудистой системы возможны кумуляция препарата или повреждение подкожной клетчатки, мышцы (при подкожном и внутримышечном введении); наконец, при инъекциях возможно инфицирование.
Следует помнить, что внутривенное введение гипертонических растворов может повредить эндотелий сосудов и нарушить функцию гистогематических барьеров.
В некоторых случаях (маленькие дети, низкое артериальное давление и т. п.) внутривенное введение затруднено. Для облегчения венепункции место инъекции за 10-15 мин до процедуры обрабатывают нитроглицериновой мазью — 0,4% (0,1 г/5 кг массы тела), а выше места венепункции устанавливают источник бестеплового света (трансиллюминация) для лучшей видимости вен.
Не рекомендуется вводить лекарственные средства в вены головы, так как при этом может произойти нарушение мозгового кровотока.
Что касается введения препаратов новорожденным в вену пуповины, следует помнить, что Аранциев (венозный) проток, через который ксенобиотик попадает в нижнюю полую вену и далее в системный кровоток, минуя печень, функционирует после рождения всего 10-15 мин (в очень редких случаях дольше – до 48 ч). При введении в более поздние сроки лекарство целиком попадает в печень и подвергается пресистемной элиминации.
Кроме осложнений, возможных при внутривенном введении, в этом случае существует опасность возникновения некроза печени.
Внутривенное введение должно быть болюсным медленным или лучше инфузионным (капельным). При введении высокоактивных препаратов и/или низких доз лекарств необходимы очень точный расчет дозы и учет величины «мертвого» объема шприца, в связи с чем лучше пользоваться разведенными растворами, кроме того, это уменьшает опасность локального повреждения эндотелия сосудов.
Таким образом, правильно выбранный путь введения обеспечивает создание оптимальной концентрации лекарства в организме и скорости наступления эффекта.
Связывание с белками плазмы крови и распределение лекарственных средств
Лекарственный препарат, попав в кровь, находится в ней в двух фракциях: свободной и связанной. Лекарства связываются, главным образом, с альбуминами, в меньшей степени — с кислыми al-гликопротеидами, липопротеинами, гамма-глобулинами и форменными элементами крови (эритроцитами). Под концентрацией лекарства в плазме крови понимают сумму свободной и связанной с белками его фракций. Особенно важно обращать внимание на связывание с белками плазмы крови, если оно превышает 70-80%, так как в некоторых случаях данный показатель может меняться. Например, связывание с белками может уменьшаться:
— при заболеваниях печени, почек, сепсисе, ожогах, белковом голодании (уменьшается синтез или увеличивается потеря белка);
— при повышении в крови уровня билирубина, остаточного азота, жирных кислот или одновременном введении нескольких препаратов (одно лекарство вытесняет другое из связи с белком);
— у недоношенных новорожденных, новорожденных и пожилых людей (онтогенетически обусловленный низкий уровень белка).
Уменьшение связанной фракции лекарства на 10-20% приводит к увеличению свободной фракции на 50-100%, что имеет особое значение при использовании препаратов с малой широтой терапевтического диапазона. Имеет значение не только процент связывания, но и степень сродства (аффинитета) ксенобиотика к белку.
Связывание с белками плазмы крови, несомненно, оказывает влияние на распределение лекарственных средств в организме. В ткани и клетки поступает только свободная фракция, именно она и оказывает фармакодинамическое действие. Однако на распределение влияют и другие факторы: степень сродства к рецептору, соотношение ионизированной и неионизированной фракции вещества, наличие лигандинов (эндогенных веществ, связывающих лекарства в клетках), относительная масса мышечной ткани, жира, внеклеточной жидкости, скорость суточной обмениваемости внеклеточной жидкости, общее содержание воды в организме и т.п.
Распределение лекарственного средства с учетом всех факторов, влияющих на этот процесс, характеризуется фармакокинетическим показателем — объемом распределения
Это — условный объем жидкости, необходимый для равно мерного распределения в нем лекарственного средства, обнаруживаемого в терапевтической концентрации в плазме крови. В большинстве руководств и справочников при характеристике лекарства приводятся величины удельного объема распределения (л/кг).
Если объем распределения меньше 0,5 л/кг, лекарственный препарат находится преимущественно в плазме крови и во внеклеточной жидкости, если больше — лекарство распределено во всей водной фазе и в маловаскуляризованных тканях.
Если объем распределения более 1 л/кг, вещество преимущественно содержится в липидах, мышцах и других тканях. В этом случае применение гемосорбции при отравлении бесполезно.
Особому правилу подчиняется проникновение лекарств в мозг, через гематоэнцефалический барьер. Гематоэнцефалический барьер — динамически функционирующая мембрана между кровью и мозгом, регулируемая самим мозгом. Через данный барьер существуют следующие виды транспорта:
— для глюкозы, аминокислот обнаружены специальные носители, синтезирующиеся эндотелием;
— для инсулина, трансферрина — специальные рецепторы, которые их захватывают, а затем интернализуются и освобождают эти вещества в интерстициальное пространство мозга;
— при соприкосновении белков плазмы крови с поверхностью эндотелиальных клеток сосудов мозга происходит конформационное изменение белка и отщепление связанного с ним вещества.
Между клетками эндотелия капилляров гипофизарной и эпифизарной областей, срединного возвышения, хориоидального сплетения и acea postrema существуют «водные поры», которые могут пропускать молекулы, имеющие массу до 30 000 дальтон.
О функции гематоэнцефалического барьера можно судить по наличию в крови специальных кислых белков. При некоторых заболеваниях мозга (менингит, травма и т.п.) проницаемость гематоэнцефалического барьера повышена.
Элиминация лекарственных средств
Элиминация — удаление лекарственного вещества из организма путем как биотрансформации, так и экскреции. Различают пресистемную и системную элиминацию.
Системная элиминация — удаление ксенобиотика после его попадания в системный кровоток.
Биотрансформация лекарств может происходить в печени, стенке кишечника, почках и других органах. Различают два этапа биотрансформации, каждый из которых может иметь и самостоятельное значение.
I этап — несинтетический (преобладает катаболическое направление реакций), идет перестройка молекул субстрата. Из лекарственных веществ путем окисления или, реже, восстановления образуются более полярные (а, значит, более гидрофильные)и менее активные метаболиты. Происходит это под влиянием монооксигеназной системы, основными компонентами которой являются цитохромы Р-450 и Р-В5, а также НАДФ (никотинамидадениндинуклеотид фосфорилированный). Однако под влиянием этой системы из ряда ксенобиотиков могут образовываться высоко реакционно-способные вещества, в том числе эпоксиды и азотсодержащие оксиды, которые при слабости обезвреживающих их систем (эпоксидгидраз, глутатионпероксидаз) способны взаимодействовать со структурными и ферментными белками и повреждать их. Они становятся чужеродными для организма и на них начинается выработка антител (аутоагрессия). Эпоксидыазотсодержащие оксиды и другие реакционно-способные метаболиты могут связываться и повреждать мембраны клеток, нарушат синтез нуклеиновых кислот, а, значит, вызывать канцерогенез, мутагенез, тератогенез.
2 этап — синтетический (анаболическая направленность реакций), образование конъюгатов с остатками различных кислот или других соединений. Образовавшиеся парные соединения фармакологически неактивны и высокополярны. Сульфатирование осуществляется в полной мере уже к рождению ребенка; метилирование — к концу 1-го месяца жизни; глюкуронидация — к концу 2-го; соединение с цистеином и глутатионом — в 3 мес, с глицином — в 6 мес. Недостаточное функционирование одного пути образования парных соединений в некоторых случаях может компенсироваться другим. Из-за незрелости ферментных систем печени в плазме крови новорожденных и грудных детей дольше остаются не подвергшиеся биотрансформации исходные жирорастворимые вещества, способные проникать в ткани и вызывать фармакологические эффекты. Вместе с тем, в печени детей этого возраста могут образовываться иные (иногда активные) метаболиты, необнаруживаемые у взрослых (например, теофиллин превращается в кофеин).
Лекарственные препараты могут влиять на скорость биотрансформации в печени, угнетая ее (индометацин, циметидин, аминазин, левомицетин, эритромицин, тетрациклин, новобиоцин, ПАСК и др.) или ускоряя (фенобарбитал, зиксорин, дифенилгидантоин (дифенин), бутадион, амидопирин, рифампицин, теофиллин, ноксирон, хлордиазепоксид и др.). Длительно назначая и/или комбинируя лекарственные препараты, необходимо учитывать такую возможность.
На биотрансформацию лекарств влияет печеночный кровоток. Если препараты (ацетилсалициловая кислота, имизин, изадрин, лидокаин, пропранолол (анаприлин), морфин, верапамил) способны быстро инактивироваться, то при остром гепатите, когда скорость кровотока не снижена (и даже может возрастать), их биотрансформация не меняется.
Она уменьшается при цирротическом процессе, с обеднением кровотока. Когда препараты (карбамазепин, дифенилгидантоин (дифенин), варфарин, дигитоксин, аминазин, хинвдин) медленно трансформируются в печени, более важна функция печеночных клеток, уровень активности ферментов которых снижался при гепатите.
Экскреция — удаление ксенобиотика из организма может быть осуществлено печенью, почками, кишечником, легкими, железа ми внешней секреции. Главное значение имеют печень и почки.
Печень экскретирует с желчью как неизмененные соединения, так и образовавшиеся в ней метаболиты. При этом большинство веществ обратно не всасываются и выводятся кишечником.
Однако глюкурониды и некоторые другие парные соединения, выделяющиеся с желчью, могут гидролизоваться кишечными или бактериальными ферментами; при этом образуются липидорастворимые вещества, которые вновь реабсорбируются и попадают в кровь, поддерживая в ней и тканях свою концентрацию, а затем вновь экскретируются с желчью. Так осуществляется энтерогепатическая циркуляция.
При печеночной недостаточности корректировка режима до зирования препаратов крайне сложна, поэтому на практике она производится эмпирически, основываясь на клинических эффектах. При невозможности модификации дозировки от препарата надо отказываться и искать ему замену.
Выведение лекарств почками складывается из их фильтрации, секреции и реабсорбции.
Фильтрация лекарств в клубочках осуществляется пассивно. Молекулярная масса веществ не должна быть больше 5-10 тыс, они не должны быть связаны с белками плазмы крови. Секреция — процесс активный (с затратой энергии при участии специаль ных транспортных систем), не зависящий от связывания препара тов с белками плазмы крови. Реабсорбция глюкозы, аминокислот, катионов и анионов происходит активно, а жирорастворимых веществ — пассивно. У детей младшего возраста (до 3 лет) эти процессы осуществляются медленнее, чем в более старшем возрасте. Способность почек к выведению лекарств путем фильтрации проверяется по экскреции эндогенного креатинина, так как оба процесса происходят параллельно с одинаковой скоростью.
Фильтрация — основной механизм экскреции почками лекарств, не связанных с белками плазмы крови. В связи с этим в фармакокинетике элиминирующую функцию почек оценивают по скорости именно этого процесса.
При почечной недостаточности корректировку режима дозирования осуществляют с помощью расчета клиренса эндогенного креатинина (С/ кр). Клиренс — это гипотетический объем плазмы крови, который полностью очищается от лекарственного средства за единицу времени. В норме клиренс эндогенного креатинина составляет 80-120 мл/мин.
Кроме того, для определения клиренса эндогенного креатини на существуют специальные номограммы. Они составлены с учетом уровня креатинина в сыворотке крови, массы тела и роста больного.
Определив клиренс, врач пользуется соответствующими рекомендациями по дозированию и/или кратности назначения соответствующего препарата.
Конечно, для контроля за коррекцией доз и режимом введения наиболее информативно определение уровня лекарств в плазме крови при известных терапевтических и токсических концентрациях вещества, но сделать это бывает не всегда возможно.
Количественно элиминацию ксенобиотика можно оценить и с помощью коэффициента элиминации. Он отражает ту часть (в процентах) лекарственного вещества, на которую происходит уменьшение его концентрации в организме в единицу времени (чаще за сутки).
Связь между объемом распределения и клиренсом вещества выражается периодом полуэлиминации (t 1/2). Период полуэлиминации вещества — это время, за которое концентрация его в плазме крови снижается наполовину. Как показатель распределения или элиминации лекарств t 1/2 играет второстепенную роль. Точное представление о величине t 1/2 не всегда подсказывает тактику введения препарата больному, так как уменьшение наполовину концентрации лекарства в плазме крови может сопровождаться как сохранением еще терапевтически действующей новой концентрации, так и возникновением уровня препарата, значительно меньшего, чем терапевтический уровень.
Известно, что при введении постоянной поддерживающей дозы препарата при одинаковом интервале дозирования в среднем через 4-5 t 1/2 в плазме крови создается его равновесная концентрация. Поэтому после начала лечения в ответ на продолжающиеся жалобы больного надо начинать реагировать через 4-5 t 1/2, т.е. надо увеличивать дозу или менять препарат. Именно через этот период можно оценивать результат терапии после отмены препарата. Наконец, исчезновение большинства нежелательных эффектов (кроме аллергических) происходит тоже в это время.
Знание и строгий учет всех вышеперечисленных фармакокинетических параметров лекарств обеспечивает сохранение их концентрации в плазме крови в пределах терапевтического диапазона.
Это имеет особо важное значение для препаратов с малой широтой терапевтического действия.
Факторы, количественно и качественно изменяющие эффект лекарств
1. Физиологические факторы:
а) возраст — дети часто более чувствительны к вызываемым лекарствами изменениям в водном и электролитном обмене, кислотно-щелочном балансе; пожилые больные могут необычно реагировать из-за нарушений распределения, инактивации и выведения лекарства вследствие возрастных анатомических и физиологических изменений в организме, а также из-за сопутствующих заболеваний;
б) пол — женщины, особенно во время беременности, могут быть более чувствительными к лекарствам;
в) хронестезия и хронергия; хропестезия — циклические изменения в чувствительности биологических систем организма к лекарствам (циркадные изменения — в течение суток; циркатригентантные — в течение месяца; цирканнуальные — в течение года); хропергия — ритмические изменения в биосистемных эффектах, в частности в эффективности лекарств; учет хронергии позволяет определять время достижения оптимального эффекта при минимальном риске побочных эффектов, например гормональных препаратов.
2. Особенности индивидуальной фармакокинетики лекарств.
3. Время приёма лекарств в зависимости от приёма и характера пищи, влияния факторов внешней среды.
4. Генетические факторы, влияющие на биоусвояемость и эффективность лекарств.
5. Лекарственное взаимодействие при приёме нескольких лекарств.
6. Сопутствующие патологические изменения в органах (печень, почки, желудочно-кишечный тракт).
7. Чувствительность больного к лекарству.
8. Приверженность больного назначаемому врачом лечению.
Таким образом, незнание фармакокинетики и фармакодинамики лекарственных средств может повлечь за собой серьезные отклонения и отягощающие состояние больного человека.
1. Лепахин В.К. Клиническая фармакология и фармакотерапия М. 1997. С. 24-42.
2. В.И. Метелица Справочник по клинической фармакологии сердечно сосудистых лекарственных средств. М.: Издательство Медицина. 1996 с. 16-33.
3. И.Б. Михайлов Клиническая фармакология и терапия 1998, С.-Пб.: с 6-30.
4. Гаевый М.Д. Фармакотерапия с основами клинической фармакологии. Волгоград. 1996. С. 6-22.
Источник


