- Изменился перечень орфанных заболеваний, препараты для лечения которых закупаются на федеральном уровне
- Перечень лекарственных препаратов при орфанных заболеваниях
- Орфанные препараты — новая категория на фармрынке
- Нередкие редкие (орфанные) заболевания
- Редкие болезни известных людей
- Примеры списка орфанных заболеваний
- Орфанные заболевания
Изменился перечень орфанных заболеваний, препараты для лечения которых закупаются на федеральном уровне
 |
| bogdan.hoda / Depositphotos.com |
На федеральный уровень передано финансирование лекарственного обеспечения граждан, страдающих неуточненной апластической анемией, наследственным дефицитом факторов II (фибриногена), VII (лабильного), X (Стюарта-Прауэра). В связи с этим Правительство РФ уточнило (Постановление Правительства РФ от 5 июня 2020 г. № 829):
- перечень жизнеугрожающих и хронических прогрессирующих редких (орфанных) заболеваний, приводящих к сокращению продолжительности жизни граждан или их инвалидности;
- правила формирования перечней лекарств и минимального ассортимента препаратов, необходимых для оказания медпомощи;
- порядок функционирования единой государственной информационной системы в сфере здравоохранения;
- программу госгарантий бесплатного оказания гражданам медпомощи на 2020 г. и плановый период 2021 и 2022 гг.
Все важные документы и новости о коронавирусе COVID-19 – в ежедневной рассылке Подписаться
Источник
Перечень лекарственных препаратов при орфанных заболеваниях
к распоряжению Правительства
от 26 декабря 2015 г. N 2724-р
ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ, ПРЕДНАЗНАЧЕННЫХ
ДЛЯ ОБЕСПЕЧЕНИЯ ЛИЦ, БОЛЬНЫХ ГЕМОФИЛИЕЙ, МУКОВИСЦИДОЗОМ,
ГИПОФИЗАРНЫМ НАНИЗМОМ, БОЛЕЗНЬЮ ГОШЕ, ЗЛОКАЧЕСТВЕННЫМИ
НОВООБРАЗОВАНИЯМИ ЛИМФОИДНОЙ, КРОВЕТВОРНОЙ И РОДСТВЕННЫХ
ИМ ТКАНЕЙ, РАССЕЯННЫМ СКЛЕРОЗОМ, ЛИЦ ПОСЛЕ ТРАНСПЛАНТАЦИИ
ОРГАНОВ И (ИЛИ) ТКАНЕЙ
Анатомо-терапевтическо-химическая классификация (АТХ)
I. Лекарственные препараты, которыми обеспечиваются больные гемофилией
кровь и система кроветворения
витамин K и другие гемостатики
факторы свертывания крови
фактор свертывания крови VIII
фактор свертывания крови IX
фактор свертывания крови VIII + фактор Виллебранда
эптаког альфа (активированный)
II. Лекарственные препараты, которыми обеспечивают больные муковисцидозом
противокашлевые препараты и средства для лечения простудных заболеваний
отхаркивающие препараты, кроме комбинаций с противокашлевыми средствами
III. Лекарственные препараты, которыми обеспечиваются больные гипофизарным нанизмом
гормональные препараты системного действия, кроме половых гормонов и инсулинов
гормоны гипофиза и гипоталамуса и их аналоги
гормоны передней доли гипофиза и их аналоги
соматропин и его агонисты
IV. Лекарственные препараты, которыми обеспечиваются больные болезнью Гоше
пищеварительный тракт и обмен веществ
другие препараты для лечения заболеваний желудочно-кишечного тракта и нарушений обмена веществ
другие препараты для лечения заболеваний желудочно-кишечного тракта и нарушений обмена веществ
V. Лекарственные препараты, которыми обеспечиваются больные злокачественными новообразованиями лимфоидной, кроветворной и родственных им тканей (хронический миелоидный лейкоз, макроглобулинемияВальденстрема, множественная миелома, фолликулярная (нодулярная) неходжкинскаялимфома, мелкоклеточная (диффузная) неходжкинскаялимфома, мелкоклеточная с расщепленными ядрами (диффузная) неходжкинскаялимфома, крупноклеточная (диффузная) неходжкинскаялимфома, иммунобластная (диффузная) неходжкинскаялимфома, другие типы диффузных неходжкинскихлимфом, диффузная неходжкинскаялимфома неуточненная, другие и неуточненные типы неходжкинскойлимфомы, хронический лимфоцитарный лейкоз)
противоопухолевые препараты и иммуномодуляторы
Источник
Орфанные препараты — новая категория на фармрынке
Роза Ягудина о росте количества орфанных препаратов и их роли в лечении пациентов
Нередкие редкие (орфанные) заболевания
Редкие болезни известных людей
Примеры списка орфанных заболеваний
Почему так дорого?
- Всероссийское общество гемофилии
- Общероссийская общественная организация инвалидов — больных рассеянным склерозом
- Межрегиональная общественная организация «Помощь больным муковисцидозом»
- Межрегиональная общественная организация «Содействие инвалидам с детства, страдающим болезнью Гоше, и их семьям»
- Региональная общественная организация помощи больным несовершенным остеогенезом
- «Хрупкие дети»
- Межрегиональная общественная организация «Содействие больным саркомой»
- Ассоциация нервно-мышечных болезней «Надежда»
- Благотворительный фонд «Подари жизнь»
Материалы об орфанных препаратах и заболеваниях:
Системные васкулиты как пример редких, орфанных заболеваний
Первичные системные васкулиты — редкие (орфанные) заболевания, и врачи, работающие в обычных лечебных учреждениях, а не в специализированных центрах, просто не могут накопить необходимого опыта в распознавании и лечении этих болезней.

Новые открытия и актуальные проблемы в мире лекарств
Сегодня орфанным препаратам стали уделять повышенное внимание. Появились препараты для лечения таких редких болезней, как мукополисахаридоз II типа, болезнь Ниманна-Пика и так далее.

Оригинальные лекарственные препараты и дженерики
В «лекарственном портфеле» многих инновационных компаний присутствуют орфанные препараты, которые окупаются долго и не приносят большой прибыли. Их разработка и производство являются следствием осознания ответственности перед каждым пациентом в отдельности.
Нашли ошибку? Выделите текст и нажмите Ctrl+Enter.

19 августа 2021

Редакция не несет ответственности за информацию, размещенную в рекламных материалах. Мнение редакции может не совпадать с мнением наших авторов. Все материалы, опубликованные в журнале, охраняются законом «Об авторском праве». Любое воспроизведение статей, перепечатка либо ссылка на них допускаются исключительно с письменного согласия редакции.
Источник
Орфанные заболевания
Один из миллиона, особенный, уникальный –
эти эпитеты подойдут гениям и тем, кому повезло меньше, –
людям с редкими болезнями.
Им не помогают обычные лекарства, они редко доживают до старости
и часто выглядят не так, как другие люди.
Мы уже неоднократно обращались к теме орфанных заболеваний. Актуальность обсуждения данной проблемы обусловлена повышенным вниманием к ней не только в России, но и во всём мире. Связано это в основном с высоким уровнем экономического и социального бремени редких болезней.
Напомним, что орфанными считаются заболевания, затрагивающие небольшую часть популяции. Не существует единого, широко принимаемого определения редких заболеваний. Некоторые определения полагаются на количество людей, живущих с заболеванием, другие могут включать иные факторы, например, доступность лечения болезни или возможность облегчения ее течения. В США Акт о редких заболеваниях (Rare Disease Act) 2002 года определяет редкие болезни как «болезни или состояния, затрагивающие менее 200 000 людей в США» («any disease or condition that affects less than 200,000 persons in the United States»), или примерно 1 человека из 1500. Низкий уровень в популяции обычно соответствует менее чем 1 из 2000. Болезни, не являющиеся угрожающими жизни, серьёзными хроническими, либо адекватно излечимыми, исключаются из определения. В России редкими предлагается считать заболевания с «распространенностью не более 10 случаев на 100 000 человек». В настоящее время в нашей стране Постановлением Правительства Российской Федерации от 26.04.2012 г. № 403 утвержден перечень из 24 редких (орфанных) заболеваний.
Таблица № 1
Перечень
жизнеугрожающих и хронических прогрессирующих редких (орфанных) заболеваний, приводящих к сокращению
продолжительности жизни граждан или их инвалидности
(утв. постановлением Правительства РФ
от 26 апреля 2012 г. № 403)
Заболевание
Код
заболевания
Пароксизмальная ночная гемоглобинурия (Маркиафавы-Микели)
Апластическая анемия неуточненная
Наследственный дефицит факторов II (фибриногена), VII (лабильного), X (Стюарта-Прауэра)
Идиопатическая тромбоцитопеническая
пурпура (синдром Эванса)
Дефект в системе комплемента
Преждевременная половая зрелость
центрального происхождения
Нарушения обмена ароматических
аминокислот (классическая фенилкетонурия, другие виды гиперфенилаланинемии)
Болезнь «кленового сиропа»
Другие виды нарушений обмена аминокислот
с разветвленной цепью (изовалериановая
ацидемия, метилмалоновая ацидемия,
пропионовая ацидемия)
Нарушения обмена жирных кислот
Другие сфинголипидозы:
болезнь Фабри (Фабри-Андерсона),
Нимана-Пика
Мукополисахаридоз, тип I
Мукополисахаридоз, тип II
Мукополисахаридоз, тип VI
Острая перемежающая (печеночная) порфирия
Нарушения обмена меди (болезнь Вильсона)
Легочная (артериальная) гипертензия
(идиопатическая) (первичная)
Юношеский артрит с системным началом
Примерно половина орфанных заболеваний обусловлена генетическими отклонениями. Симптомы могут быть очевидны с рождения или проявляться в детском возрасте. Проблема редких заболеваний особенно актуальна для педиатрии, так как 2/3 редких болезней манифестируют в раннем возрасте. Реже встречаются токсические, инфекционные или аутоиммунные «сиротские» болезни. Причинами их развития могут быть наследственность, ослабление, плохая экология, высокий радиационный фон, у мамы и у самих детей в раннем возрасте. Большинство орфанных заболеваний – хронические. Они в значительной мере ухудшают качество жизни человека и могут стать причиной летального исхода. Для большинства таких болезней не существует эффективного лечения. Основа терапии таких больных – улучшение качества и увеличение продолжительности жизни пациентов.
Впервые на территории Российской Федерации орфанные заболевания получили юридический статус в 2011 году с принятием 21 ноября2011 г. Федерального закона № 323-ФЗ «Об основах охраны здоровья граждан в Российской Федерации». В соответствии с ч. 9 ст. 83 данного федерального закона граждане РФ, страдающие жизнеугрожающими и хроническими прогрессирующими редкими (орфанными) заболеваниями, имеют право на обеспечение препаратами, предназначенными для лечения этих заболеваний, за счет средств субъектов РФ. Однако большинство региональных бюджетов орфанной нагрузки не выдерживают и даже ограниченный перечень из 24 редких заболеваний обслуживают с трудом [3].
На сегодняшний день на территории Брянской области проживают 107 пациентов, страдающих редкими (орфанными) заболеваниями, из них 55 детей. Для обеспечения указанной категории пациентов необходимо 300 миллионов рублей в год, дефицит финансирования программы в регионе составляет 87%.
Таблица № 2
Количество пациентов Брянской области,
страдающих редкими (орфанными) заболеваниями
Заболевание
Численность
Апластическая анемия неуточненная (D61.9)
Гемолитико-уремический синдром (D59.3)
Дефект в системе комплемента (D84.1)
Идиопатическая тромбоцитопеническая пурпура (синдром Эванса) (D69.3)
Мукополисахаридоз, тип VI (E 76.2)
Нарушения обмена ароматических аминокислот (классическая фенилкетонурия, другие виды
гиперфенилаланинемии) (Е70.0, Е70.1)
Нарушения обмена меди (болезнь Вильсона)
(E 83.0)
Наследственный дефицит факторов
II (фибриногена), VII (лабильного),
Х (Стюарта-Прауэра) (D68.2)
Незавершенный остеогенез (Q78.0)
Пароксизмальная ночная гемоглобинурия
(Маркиафавы-Микели) (D59.5)
Юношеский артрит с системным началом
(M 08.2)
Стоимость лечения пациентов с различными редкими заболеванием различная. Например, для лечения галактоземии необходимо специальное лечебное питание: без галактозы и лактозы. Стоимость его лишь незначительно отличается от стоимости обычного питания для детей. Но лечение других пациентов может составлять сотни тысяч в месяц. При составлении Перечня не учитывалась стоимость лечения и с общечеловеческой точки зрения, включение заболеваний в перечень вне зависимости от стоимости лекарства подчёркивает равные права больных в доступности современного лечения.
Депутаты государственной Думы, органы исполнительной власти субъектов РФ и общественные пациентские организации неоднократно поднимали вопрос о необходимости привлечения средств федерального бюджета к софинансированию региональных программ обеспечения лекарственными препаратами лиц, страдающих редкими (орфанными) заболеваниями. Проблемы финансирования программ в регионах усугубляется отсутствием единой модели оказания медицинской помощи, в том числе лекарственными препаратами, данной категории граждан. Каждый субъект РФ решает эти проблемы по-своему. При этом есть субъекты, в которых ориентированность на сохранение жизни пациента взята за основу при принятии решений по лечению больных с редкими заболеваниями (республики Башкоркостан, Дагестан, Коми, Калмыкия, Московская область и другие). В других же (республика Татарстан, Нижегородская область) добиться обеспечения лекарственными препаратами возможно только через судебные решения, которые также не сразу исполняются региональными властями.
Еще около 30 лет назад препаратов, предназначенных для лечения орфанных заболеваний, практически не было. Из-за узости рынка большинство фармкомпаний разработку и лонч таких лекарственных средств (ЛС) считали непривлекательными. По версии Tufts Center, чтобы вывести орфанный препарат на рынок США, компания должна потратить не менее 1,2 млрд дол. Ограниченное число пациентов для проведения клинических исследований лишь усугубляло ситуацию. В 1983 году в США был издан Orphan Drug Act, в котором впервые появилось понятие «орфанные заболевания». В документе прописывались преференции для производителей орфанных препаратов, в том числе налоговые льготы в размере 50% на проведение клинических испытаний на территории США, упрощение процедуры лицензирования и сокращение стоимости регистрации, предоставление эксклюзивных прав на продажу сроком на семь лет.
В 90-е годы аналогичные документы были изданы в Сингапуре, Японии и Австралии, в 2000 году – в Европе.
Благодаря этим мерам количество орфанных ЛС возросло. По прогнозам EvaluatePharma, мировой объем продаж препаратов для лечения редких болезней с 90 млрд дол., зафиксированных в 2013 году, через пять лет подберется к отметке в 127 млрд дол. Среднегодовой темп роста сегмента составит 7,4%, что вдвое выше аналогичного показателя для Rx-препаратов.
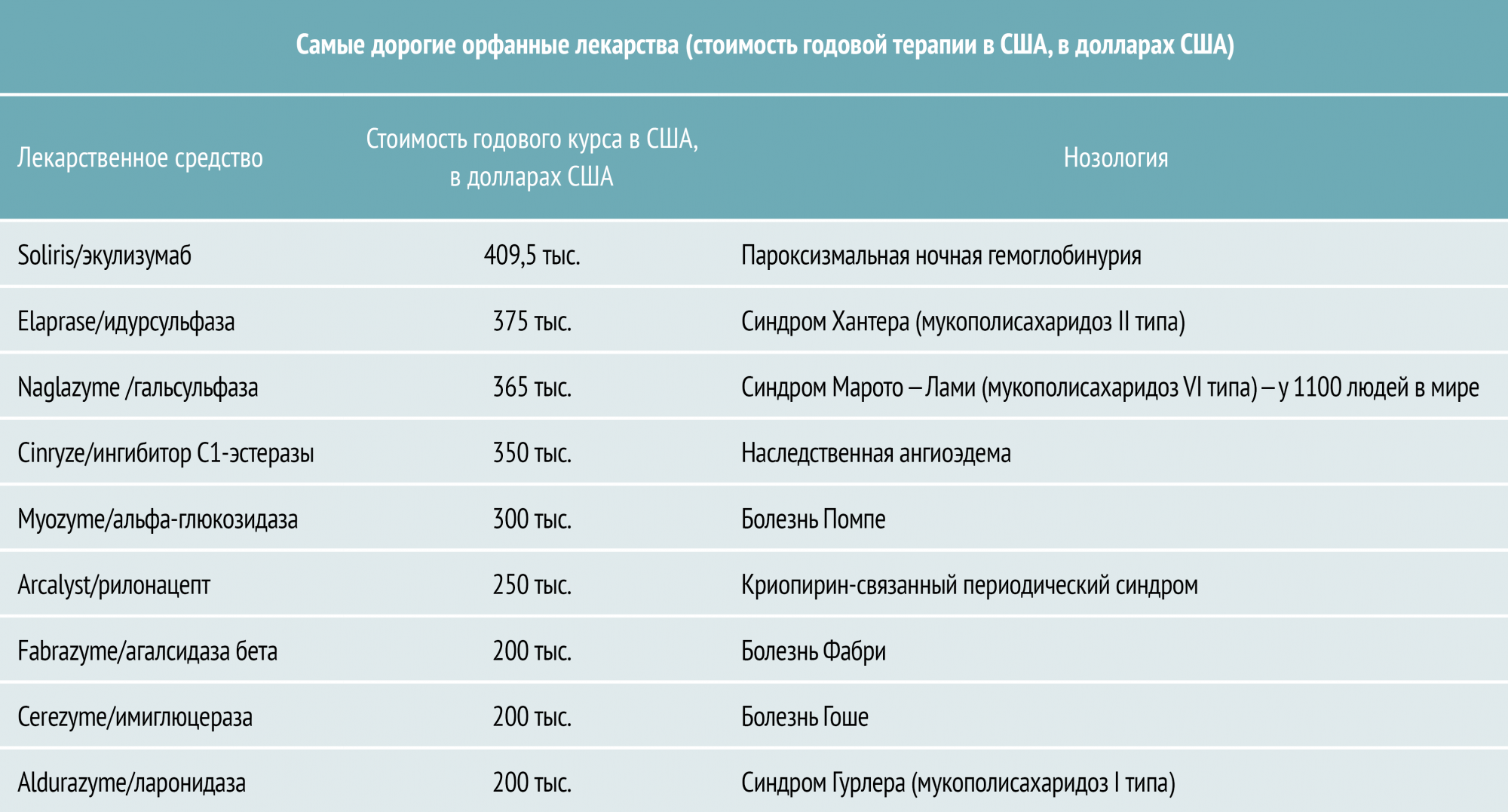
На сегодняшний день самой дорогостоящей нозологией среди входящих в Перечень, является пароксизмальная ночная гемоглобинурия. Пароксизмальная ночная гемоглобинурия (ПНГ) – редко встречающееся приобретенное заболевание, вызванное нарушением эритроцитарной мембраны и характеризующееся хронической гемолитической анемией, перемежающейся или постоянной гемоглобинурией и гемосидеринурией, явлениями тромбоза и гипоплазией костного мозга. Это заболевание обычно впервые диагностируется у лиц в возрастной группе 20–40 лет, но может встречаться и у пожилых. При классической форме гемолиз (разрушение эритроцитов крови с выделением в окружающую среду гемоглобина) происходит в то время, когда больной спит (ночная гемоглобинурия), что может быть обусловлено небольшим снижением ночью рН крови. Однако гемоглобинурия (появление гемоглобина в моче.) наблюдается только примерно у 25% больных, причем у многих не в ночное время. Препарат, использующийся для лечения данного заболевания, – самый дорогостоящий в мире. Годовой курс лечения препаратом Солирис Soliris (МНН: экулизумаб) от швейцарской компании Alexion Pharma оценивается в 400 тыс. дол.
Elaprase (идурсульфаза, «Shire Human Genetic Therapies, Inc.») компенсирует недостаток одного из лизосомальных ферментов, генетически обусловленная нехватка которого развивается при синдроме Хантера (мукополисахаридозе II типа). Синдром Хантера – одна из форм мукополисахаридоза, возникает в результате дефицита ряда ферментов, что приводит к накоплению белково-углеводных комплексов и жиров в клетках. Болезнь проявляется в раннем возрасте (2–4 года) утолщением ноздрей, губ, языка,
тугоподвижностью суставов, задержкой роста. До двух лет отмечают такие признаки, как шумное дыхание (обструкция верхних дыхательных путей), риниты, паховые и пупочные грыжи. До появления Elaprase лечение синдрома Хантера ограничивалось паллиативной терапией. В России препарат зарегистрирован в 2008 году, стоимость лечения составляет 350 тыс. дол./год.
Naglazyme (гальсульфаза, «BioMarin Pharmaceuticals») – первый и единственный препарат для лечения синдрома Марото–Лами (мукополисахаридоза VI типа). Naglazyme замещает недостающий при этом заболевании фермент (арилсульфатаза). Заболевание характеризуется отложением дерматансульфата в различных тканях. Манифестирует к 2–3 годам жизни отставанием в росте, огрублением черт лица, напоминающим фенотип синдрома Гурлер, помутнением роговицы, деформациями скелета (поясничный кифоз, вальгусное искривление голеней), гепатоспленомегалией, тугоподвижностью суставов, грыжами. Умственная отсталость не характерна. Пациенты, принимающие Наглазим, смогли преодолеть больше ступенек лестницы и ходить на более далекие расстояния, чем раньше. Синдром Марото–Лами отмечен в настоящее время примерно у 1100 людей в мире. Стоимость лечения составляет около 350 тыс. дол./год.
Ещё один дорогостоящий препарат – Myozyme (альглюкозидаза альфа, «Genzyme Corporation») показан для лечения пациентов с болезнью Помпе (дефицитом альфа-глюкозидазы, которая необходима, чтобы разрушать гликоген – вещество, которое является источником энергии для организма). Симптомы болезни Помпе возникают из-за аномального накопления гликогена в клетках. В настоящее время болезнь Помпе не входит в Перечень, однако пациентские сообщества и медицинские организации ведут активную работу по расширению Перечня-24, в частности по включению в него болезни Помпе. Без заместительной терапии дети с ранним началом заболевания умирали в течение первого года жизни вследствие недостаточности кровообращения или инфекций респираторного тракта. Согласно результатам клинических испытаний Myozyme увеличивал период выживаемости без искусственной вентиляции легких у таких пациентов. Лечение ребенка этим препаратом в течение года стоит около 100 тыс. дол., взрослого – около 300 тыс. дол.
Aldurazyme (ларонидаза, «Genzyme Corporation») предназначен для ферментозаместительной терапии при мукополисахаридозе I типа (синдром Гурлера) – тяжелом прогрессирующем, часто жизнеугрожающем, заболевании. Стоимость лечения препаратом составляет 200 тыс. дол./год [5].
Улучшить ситуацию с обеспечением лекарствами пациентов, страдающих редкими заболеваниями, должны готовящиеся в настоящее время федеральные нормативно-правовые акты, в частности поправки в закон «Об обращении лекарственных средств» №61-ФЗ: изменения, дающие преференции орфанным препаратам, подготовлены, но пока не приняты. Также решения требует принципиальный вопрос – передача финансирования помощи гражданам, страдающим редкими заболеваниями, с регионального на федеральный уровень.
С 2008 года последний день февраля объявлен Днём редких заболеваний.
Источник


