Потеряли термин


Есть случаи, когда качеством копии можно пренебречь. Но есть области, в которых качество копии определяет вопрос сохранения жизни и здоровья. Это выпуск воспроизведенных лекарств.

Согласно существующим международным регуляторным актам, такие понятие, как «оригинальное лекарственное средство» и «референтное лекарственное средство», разделены. Под термином «оригинальное» понимается лекарство с новым действующим веществом, которое было первым зарегистрировано на мировом фармацевтическом рынке на основании досье, содержащего результаты полных доклинических и клинических исследований, подтверждающих его безопасность и эффективность. Термин «референтный» возникает в момент проведения исследования биоэквивалентности и относится к лекарству, используемому в качестве препарата сравнения.
В руководствах по проведению клинических исследований биоэквивалентности допускаются достаточно большие отличия фармакокинетических параметров двух сравниваемых лекарств. Так, их биоэквивалентность считается доказанной, если отношение сравниваемых параметров в организме человека укладывается в интервал 80,00-125,00 процента при 90-процентном доверительном интервале. В этом случае, даже при получении результатов на самой грани допустимых значений, лекарства считают «одинаковыми». То есть воспроизведенное лекарство признается эффективной и безопасной копией референтного препарата.
А есть ли у них отличия? Конечно, да. В частности, параметры фармакокинетики могут отличаться на большую величину. Это значит, что лекарства — с определенными оговорками — могут быть взаимозаменяемыми. Но если мы хотим создать копию самого первого (по мировой терминологии — оригинального) лекарственного средства, то в качестве референтного необходимо взять именно исходный, оригинальный продукт, а не его копию.
В частности, в Управлении по контролю за лекарственными средствами и продуктами питания США (Food and Drug Administration, USA) разработан общедоступный перечень взаимозаменяемых «терапевтических эквивалентов», в котором, помимо прочего, есть оценка терапевтической эквивалентности пар «оригинальное лекарство — воспроизведенное лекарство» с присвоением кодового обозначения степени терапевтической эквивалентности. При этом далеко не все воспроизведенные лекарства имеют высшую оценку терапевтической эквивалентности оригинальному средству. А ведь действующее вещество в них одно и то же. Таким образом, воспроизведенное лекарство — это копия исходного (оригинального) препарата с определенными допущениями и погрешностями.
Производя очередной воспроизведенный аналог какого-либо лекарства, в качестве референтного лучше всего брать оригинальный лекарственный препарат. Поскольку если в качестве референтного препарата будет выбран уже воспроизведенный, то будущий новый продукт будет отличаться от референтного еще на одну величину допущений.
К сожалению, сегодня в России мы не можем опираться на понятие оригинального препарата. В российском законодательстве такого термина просто нет. В действующей редакции Федерального закона «Об обращении лекарственных средств» вместо термина «оригинальный лекарственный препарат» введен термин «референтный лекарственный препарат». Под ним понимается «лекарственный препарат, который впервые зарегистрирован в России, качество, эффективность и безопасность которого доказаны на основании результатов доклинических исследований лекарственных средств и клинических исследований лекарственных препаратов и который используется для оценки биоэквивалентности или терапевтической эквивалентности, качества, эффективности и безопасности воспроизведенного или биоаналогового (биоподобного) лекарственного препарата». Из этого следует, что в России первым может быть зарегистрирован не оригинальный препарат, прошедший полную программу клинической и доклинической разработки, а его копия. А оригинальный препарат может быть вообще не зарегистрирован — по разным причинам. В этом случае, руководствуясь приведенным территориальным ограничением — «впервые зарегистрирован на территории РФ», — мы должны будем при проведении исследования биоэквивалентности сравнивать новый отечественный воспроизводимый препарат с уже воспроизведенным. То есть будем делать копию с копии, не будучи уверенными, что данные по эффективности и безопасности, полученные при клинических исследованиях оригинального препарата применимы по отношению к вновь воспроизводимому.
Возможно, этот факт кто-то сочтет несущественным — ведь можно подобрать эффективную дозу и для дженерика, а затем ее корректировать. Но это не всегда так. И в первую очередь это касается вопросов мониторинга безопасности. В этом случае отсутствие понятия «оригинальный лекарственный препарат» создает целый комплекс затруднений — как административных, так и практических.
К примеру, в 2013 году Росздравнадзор выпустил методические рекомендации по подготовке периодических отчетов по безопасности. В них указано, что отсчет срока подготовки отчета должен вестись от даты первой регистрации препарата с данным действующим веществом в мире, а не на территории какой-либо страны. Оценка рисков и сигналов по данному действующему веществу в мире в этом случае синхронизирована, а регуляторные агентства будут одновременно получать информацию и иметь полноценные базы данных.
А в приказе ведомства № 1071 от 15 февраля 2017 г. указано, что порядок исчисления даты окончания сбора сведений для очередного периодического отчета утверждается самим Росздравнадзором. И лишь для лекарств, сроки и периодичность представления отчета на которые не утверждены Росздравнадзором, срок отсчитывается от даты первой регистрации в мире. Таким образом, синхронизация с мировыми сроками переоценки пользы-риска применения конкретного препарата может отсутствовать.
Почему так важна синхронизация сроков? Во-первых, синхронизация отчетов всех производителей лекарств одного международного непатентованного названия позволяет регуляторным органам одновременно получить от всех производителей все данные по безопасности, которые были ими собраны за завершенный отчетный период. Чем больше данных, тем более объективную оценку дает регуляторный орган, так как основные статистические методы глобальной обработки данных по безопасности построены на статистических методиках выявления диспропорций. А в случае асинхронности поступления информации мощности статистического метода может не хватить для того, чтобы выявить тот или иной сигнал. Изолируясь от этой системы, регуляторные органы России искусственно занижают как свои, так и глобальные возможности по своевременной идентификации сигналов по безопасности, создавая риски не только себе, но и мировому сообществу в целом. Кроме того, хотя мониторинг безопасности лекарств — процесс непрерывный, оценка безопасности препаратов на глобальном уровне происходит с определенной периодичностью. Отсутствие синхронности в предоставлении отчетов по безопасности может привести к тому, что некоторые риски, связанные с очень редкими или отсроченными в развитии нежелательными явлениями, могут быть выявлены с опозданием, что также создает довольно масштабную угрозу.
Помимо этого, отличия параметров фармакокинетики, обусловленные особенностями проведения клинических исследований биоэквивалентности, ведут к тому, что из-за искусственно полученных «слишком больших» или «слишком малых дозировок» по сравнению с оригинальным лекарством, возникает благоприятная почва для токсических эффектов либо для случаев отсутствия эффективности.
Особенно наглядно это видно на психотропных препаратах, обладающих узким терапевтическим диапазоном. При снижении дозы могут возникнуть опасные для жизни судороги, а при увеличении — еще и галлюцинации, которые не только создают угрозу для жизни больных, но и значительно снижают качество жизни. В то же время при правильно подобранной фармакотерапии больные могут длительно сохранять не только качество жизни, но и трудоспособность.
Отмеченный разброс в параметрах фармакокинетики создает условия для искусственно созданных проблем в профиле безопасности — отсутствия эффективности с одновременным токсическим воздействием. Это создает ложное впечатление «неблагополучности» данного действующего вещества, что закономерно и необратимо снижает приверженность лечению у больных и веру в препарат у врачей. Причем эта вера — как субъективный фактор и постмаркетинговые исследования эффективности — как фактор объективный могут привести к тому, что вероятно эффективная и безопасная молекула с высоким терапевтическим потенциалом окажется отозванной с рынка при очередном сеансе глобальной оценки пользы-риска. Отзыв с рынка — не самое редкое явление. За годы активного развития систем фармаконадзора как на глобальном, так и на локальном уровне накопилось немало примеров отзывов лекарства с рынка из-за того, что они не оправдали ожиданий, выйдя за пределы контролируемых условий клинических исследований. Так годы упорного труда, огромные человеческие и финансовые инвестиции, а главное — надежда пациентов на возможности современной медицины могут быть обесценены лишь по причине исключения из закона фундаментального понятия «оригинальный препарат». И это лишь начало.
Евгений Рогов, независимый GCP/GLP аудитор:
— Думаю, что в данном случае все произошло по тому же принципу «Хотели как лучше, а получилось как всегда». Изначально список референтных препаратов создавался для определения взаимозаменяемости (что не одно и то же). При этом во всем мире дженериком считается препарат по отношению к оригинальному, а в России — к первому зарегистрированному на рынке. Получается парадокс: оригинальный препарат, зарегистрированный в России позже, чем его копия, для России будет дженериком к своему дженерику.
Допустимое отличие между оригинальным препаратом и копией составляет 20 процентов. Предположим, в России впервые был зарегистрирован дженерик, который по своим параметрам отличался от оригинального лекарства на 14 процентов. Если взять такой препарат в качестве оригинального и сравнить его с другим дженериком, который тоже будет отличаться от него на 18 процентов, то итоговое отличие второго дженерика от оригинального препарата составит уже 32 процента. А эти отличия существенно влияют на безопасность и эффективность лекарств. Промахи на этапе разработки (в том числе и с выбором референтного препарата) с момента его государственной регистрации и выхода на рынок, создают риски для жизни и здоровья неопределенного круга лиц.
Оксана Монж, генеральный менеджер рецептурного подразделения компании «Санофи» в России и Беларуси:
— Для препаратов, признанных референтными или воспроизведенными, мы видим потенциальное влияние на методику расчета предельных отпускных цен производителей при включении препаратов в Перечень ЖНВЛП. В проекте перечня таких препаратов у ряда МНН отсутствуют указания на торговые наименования, что может привести к значительным сложностям в процессе регистрации цен. Кроме того, и для государственных и муниципальных заказчиков, и для участников закупок очень важно своевременное завершение работы экспертной организации Минздрава России по проведению экспертизы на взаимозаменяемость для всех лекарственных препаратов, обращающихся на рынке. Для нас остается неясным вопрос, каким образом будет определяться взаимозаменяемость тех препаратов, которые давно обращаются на рынке и по которым не планируется самостоятельно запрашивать этот статус. Нужно ли нам предоставлять необходимую для этого информацию, без которой, как мы понимаем, экспертная организация не может определить статус взаимозаменяемости. Для проведения всех аукционных процедур в полном соответствии с требованиями законодательства не менее важна и постоянная актуализация этих данных по мере появления новых продуктов или изменения статуса уже зарегистрированных препаратов.
Источник
Генерики и оригинальные препараты: взгляд практического врача
*Импакт фактор за 2018 г. по данным РИНЦ
Читайте в новом номере
Всемирная Организация Здравоохранения (ВОЗ) в 2004 г. приняла резолюцию, провозгласившую радикальное увеличение безопасности лечения приоритетной задачей ВОЗ [1]. В этой резолюции особый акцент сделан на право больного знать все о своем заболевании, методах его лечения и на необходимости получения информированного согласия больного на лечение. В связи с этим следует учитывать, что информированное согласие предполагает заведомое разъяснение пациенту различий между «аналогами» лекарственных средств (ЛС). Однако только 60% врачей обнаруживают знакомство с такой постановкой вопроса [2].
Проблема генерических препаратов чрезвычайно актуальна для России. Во–первых, потому что в РФ – самая большая доля генериков на фармацевтическом рынке (78–95%) (рис. 1). Некоторые оригинальные ЛС имеют огромное количество генериков. Так, у Вольтарена – 120 генерических «копий», что само по себе противоречит международной практике. Обычно же на фармацевтическом рынке присутствует 4–5 генериков, качество которых скрупулезно проверено при регистрации препаратов.
Во–вторых, потому что генерики, производимые в РФ, в большинстве своем – низкого качества. Так, Сергей Иванов, первый заместитель Председателя Правительства РФ на встрече с членами Общественной палаты заявил: «Я не могу не упомянуть проблему фармакологической промышленности. Наряду с лесной и рыбной отраслями, я их отношу к самым криминализированным в экономике России. Ведь там и контрафакта полно, а даже если это не контрафакт, то качество все равно низкое. Здесь давно пора навести порядок» [3].
Так чем же генерический препарат отличается от оригинального?
Оригинальный лекарственный препарат – это впервые синтезированное, прошедшее полный цикл доклинических и клинических исследований ЛС, защищенное патентом на срок до 20 лет. Только по истечении срока действия патента возможно воспроизведение ЛС любой фармацевтической компанией: создается генерик. Таким образом, генерик должен появляться значительно позже оригинального препарата. Однако уже на этом этапе в РФ имеют место нарушения. Например, патент на оригинальный азитромицин истек только в 2007 г.(!!), а в РФ уже зарегистрировано не менее 14 генериков [4].
Термин «генерик» возник в 70–е годы XX века, когда считалось, что препараты–аналоги надо называть родовым (генерическим) именем, в отличие от оригинального ЛС, которое продавалось под специальным торговым наименованием. Смысл этого правила заключался в том, что оно облегчало распознавание оригинального ЛС среди препаратов–аналогов. В настоящее время генерики часто имеют собственные названия, что не позволяет различать генерики и оригинал. Например, оригинальный препарат диклофенака называется Вольтарен (Новартис), а генерики, которые теоретически должны называться родовым (генерическим) именем диклофенак, имеют самые разнообразные наименования.
В целом основными признаками генерика должны являться:
• отсутствие патентной защиты;
• сравнительно низкая цена;
• назначение и продажа под непатентованным наименованием (МНН);
• почти полное соответствие оригинальному продукту по составу (вспомогательные вещества могут быть иными);
• соответствие фармакопейным требованиям;
• производство в условиях GMP (надлежащая производственная практика).
Однако в РФ генерики сегодня это:
• сравнительно низкая цена – не всегда;
• назначение и продажа под непатентованным наименованием (МНН) – далеко не всегда;
• почти полное соответствие оригинальному продукту по составу (вспомогательные вещества могут быть иными) – большие проблемы;
• соответствие фармакопейным требованиям – не всегда;
• производство в условиях GMP – не всегда.
В законе РФ «О лекарственных средствах» (1998) введено определение понятия «воспроизведенные лекарственные средства», однако в нем упущен центральный элемент: не указано, что они являются копиями или аналогами оригинальных препаратов [1].
Согласно международному стандарту генерик – это лекарственный продукт с доказанной фармацевтической, биологической и терапевтической эквивалентностью с оригиналом.
Фармацевтическая эквивалентность
Фармацевтическая эквивалентность предполагает содержание одних и тех же активных субстанций в одинаковом количестве и в одинаковой лекарственной форме; соответствие требованиям одних и тех же или сходных стандартов [5]. «Фармацевтически альтернативными являются лекарства, содержащие одинаковый терапевтический компонент, представленный в виде различных солей, эфиров или комплексов, либо в различных дозах» [6].
К сожалению, в РФ отменен аналитический контроль генерических препаратов на этапе регистрации, он проводится только для новых отечественных ЛС [7].
Биологическая эквивалентность
«Биоэквивалентные лекарственные препараты – это фармацевтически эквивалентные или фармацевтически альтернативные препараты, обладающие сопоставимой биодоступностью, изученной в сходных экспериментальных условиях. Под биодоступностью понимается скорость и доля всасывания активного ингредиента или активного компонента лекарства, которое начинает действовать в точке приложения» [6].
В сущности, биоэквивалентность – это эквивалентность скорости и степени всасывания оригинала и генерика в одинаковых дозах по концентрации в жидкостях и тканях организма. Тест проводится с участием 18–24 здоровых добровольцев, которые однократно принимают 1 дозу изучаемого генерика (для пролонгированного препарата – 5 суток приема), затем после нескольких периодов полувыведения принимают аналогичную дозу оригинального препарата. При сравнении двух графиков фармакокинетики различия не должны превышать 20% (рис. 2). Исследование на биоэквивалентность обязательно проводится для всех генерических препаратов. Естественно, биоэквивалентность – это не гарантия, а предположение терапевтической эквивалентности и безопасности препарата [8].
Если генерик разрешен к применению в других странах, он регистрируется в РФ по упрощенной схеме (без определения биоэквивалентности). Только генерики новых производителей исследуются на биоэквивалентность. Из 1256 зарегистрированных в 2001 г. зарубежных препаратов только 22 прошли экспертизу на биоэквивалентность при регистрации в РФ. Таким образом, при регистрации зарубежных генериков в РФ мы полностью доверяем досье, представляемым фармацевтическими компаниями [2]. Такая «доверчивость» дорого обходится пациентам. Так, по мнению директора Государственного фармакологического центра МЗ Украины В.Т. Чумака – «…ни один из 150 имеющихся на рынке Украины препаратов диклофенака по своим фармакокинетическим свойствам не соответствует Вольтарену» [9].
Хорошо известен пример контрольной проверки биоэквивалентности генериков оригинальному кларитромицину. Работа C.N. Nightingale была представлена в 2000 г. на 5–й Конференции по макролидам, азалидам, стрептограминам, кетолидам и оксазолидинонам в Севилье [10]. Автор сравнил оригинальный препарат кларитромицина с 40 копиями в отношении биоэквивалентности, применив стандарты Американской фармакопеи. Исследование показало, что 70% генериков растворяются значительно медленнее оригинального препарата, что критично для их усвоения. 80% генериков отличаются от оригинала по количеству действующего начала в одной единице продукта. Количество примесей, не имеющих отношения к действующему началу, в большинстве образцов больше, чем в оригинале. В «лучшем» генерике их было 2%, в «худшем» – 32%.
Терапевтическая эквивалентность
«Терапевтически эквивалентными лекарственные препараты могут считаться только в том случае, если они фармацевтически эквивалентны и можно ожидать, что они будут иметь одинаковый клинический эффект и одинаковый профиль безопасности при введении пациентам в соответствии с указаниями в инструкции» [6].
Генерик терапевтически эквивалентен другому препарату, если он содержит ту же активную субстанцию и, по результатам клинических исследований, обладает такой же эффективностью и безопасностью, как и препарат сравнения, чья эффективность и безопасность установлены.
Надо отметить, что сравнительное исследование должно проводиться по определенным правилам (GCP – надлежащая клиническая практика) и должно быть:
• независимым;
• многоцентровым;
• рандомизированным;
• контролируемым;
• длительным (средняя продолжительность приема ЛС);
• с жесткими конечными точками (смерть, инфаркт, инсульт).
Такого рода исследования выполнены только для единичных генериков, применяемых в РФ. К сожалению, данные клинических исследований при регистрации в РФ обязательны только для новых оригинальных препаратов, эффективность и безопасность генериков остается без внимания [7].
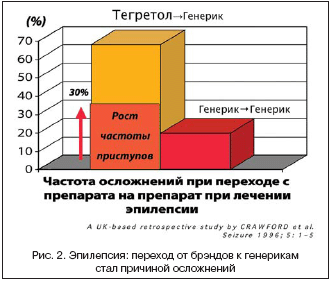
Отсутствие исследований на терапевтическую эквивалентность при регистрации генериков имеет многочисленные негативные последствия. Так, в хорошо известном исследовании противоэпилептических препаратов было показано, что переход с оригинального ЛС Тегретола на генерик приводил в 70% случаев к негативным последствиям, а в 30% случаев к значимому повышению частоты приступов [11].
По мнению профессора В.А. Насоновой, «…распространение в РФ огромного количества генериков диклофенака, не всегда обладающих основными качествами оригинального ЛС, стали заметно влиять на авторитет Вольтарена» [12].
Примером отсутствия терапевтической эквивалентности являются результаты сравнительного изучения антигипертензивной эффективности генериков эналаприла и оригинального ЛС – Ренитека. Продемонстрировано, что дозе Ренитека 12 мг, необходимой для достижения целевого АД, были эквивалентны различные дозы генериков [13].
Таким образом, особенностями генериков в РФ являются:
• появление многих генериков в России раньше соответствующих оригинальных ЛС;
• отсутствие данных о терапевтической эквивалентности с оригинальным ЛС;
• относительно высокие цены генериков;
• самая высокая доля генериков на фармацевтическом рынке.
Интересными являются сведения о том, из чего складывается стоимость оригинальных ЛС и генериков. 80% стоимости оригинального ЛС составляет стоимость исследований эффективности и безопасности препарата, а 20% стоимости – это стоимость синтеза лекарственного вещества. Вообще процесс создания оригинального ЛС очень длительный и дорогостоящий. Сначала создается молекула, потом она оценивается в исследованиях на клетках и тканях, затем на животных. После этого следуют три этапа клинических исследований на здоровых добровольцах и пациентах. После завершения клинических исследований ЛС проходит регистрацию.
Исследование оригинального ЛС продолжается и после регистрации. Проводятся с соблюдением правил GCP так называемые пострегистрационные исследования.
Известно, что только 1 из 5 000 молекул доходит до рынка в виде ЛС. Этот путь продолжается 12–15 лет и «стоит» от 800 млн. до 1 млрд. $. Прибыльными являются только 1–2 из вновь созданных ЛС.
Таким образом, преимуществами оригинальных ЛС являются:
• доказанная эффективность;
• доказанная безопасность;
• инновационность;
• воспроизводимость эффекта;
• жесткий контроль качества.
Процесс создания генерика происходит совершенно иначе. Отсутствуют три этапа клинических исследований, перед регистрацией проводится только исследование биоэквивалентности. 50% себестоимости составляет стоимость активной субстанции. Для того чтобы снизить стоимость, фармацевтические компании либо изменяют методы синтеза, либо ищут возможность приобретения наиболее дешевых субстанций. В целом приобретение активной субстанции у компаний, специализирующихся на ее производстве, является общепринятой практикой. Зачастую активная субстанция приобретается в странах, мало доступных для контроля: Китай, Индия, Вьетнам. Поставки субстанций происходят через большое количество посредников, сведения о месте производства обычно не публикуются и готовый продукт рекламируется, как изготовленный в высокоразвитой стране [1].
Качество наполнителей тоже имеет большое значение: любое изменение в составе вспомогательных веществ или оболочки могут существенно изменить качество препарата, его биодоступность, привести к токсическим или аллергическим явлениям. В том, что генерики отличаются от оригинала составом вспомогательных веществ, можно легко убедиться, изучив данные фармацевтических справочников.
Таким образом, источниками низкой стоимости генерических ЛС являются:
• отсутствие клинических исследований;
• отсутствие сравнительных исследований с оригиналом;
• отсутствие изучения профиля безопасности.
Могут ли генерики низкого качества повлиять на здоровье нации? Да, могут. Например, применение генерических антибиотиков приводит к хронизации заболеваний, резистентности бактерий, вирусов и грибов к антимикробным препаратам, росту инвалидизации и смертности [14].
Несоблюдение международных требований к генерикам в РФ влечет за собой огромные потери от «выгодной» цены вследствие затрат на повторные госпитализации, большие дозы ЛС и лечение побочных эффектов.
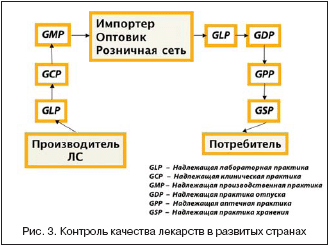
Как решается проблема генерических препаратов в развитых странах? Прежде всего это жесткий контроль качества на каждом этапе пути ЛС от производителя к пациенту (рис. 3). Кроме того, в развитых странах имеет место отрицательное отношение к биоэквивалентности, как к единственному способу оценки равнозначности лекарств. Для того чтобы считать генерик качественным, необходимо проведение клинических исследований на терапевтическую эквивалентность.
Очень важно то, что врач имеет полноценную информацию о том, какое ЛС является оригинальным препаратом, а также о качестве генерических препаратов. В США генерики разделены на группы «А» и «В». Код «А» присваивается генерикам, прошедшим клинические исследования на терапевтическую эквивалентность и имеющим отличия биоэквивалентности от оригинального ЛС не более 3–4%. Генерики с кодом «А» могут являться заменой оригинальному препарату по финансовым соображениям. Код «В» присваивается генерикам, не прошедшим клинические испытания на терапевтическую эквивалентность. Генерик с кодом «В» не может быть автоматической заменой оригинальному препарату или другому генерику с кодом «А». Сведения о статусе лекарственных препаратов – общедоступны и содержатся в справочнике «Orange Book». В аптеке провизор может отпустить больному препарат только с тем торговым названием, которое выписал врач [1,2,6].
Как решить проблему генериков в России? Конечно, наиболее кардинальным является изменение законодательства, приведение его в соответствие с законодательством цивилизованных стран. Пока этого не произошло, возможно оценивать качество генериков по некоторым весьма существенным критериям.
Во–первых, по мнению экспертов качество генерика вполне удовлетворительно, если ЛС зарегистрировано в странах с развитой контрольно–разрешительной системой, к которым относятся члены PIC–PI C/S – «Конвенции и схемы сотрудничества по фармацевтическим инспекциям» (старые члены Евросоюза, США, Япония, Канада, Австралия, Австрия, Швейцария, Норвегия, Венгрия, Чехия, Словакия, Сингапур) [1]. Важно именно место регистрации, а не производства ЛС.
Во–вторых, генерик имеет достойное качество, если компания–производитель доказала терапевтическую эквивалентность генерика оригинальному препарату в пострегистрационных рандомизированных контролируемых исследованиях [15].
В–третьих, должна быть уверенность, что при производстве генерика соблюдаются правила GMP [16].
Что делать врачу в России, если он хочет качественно лечить пациента? Скорее всего, назначать оригинальный препарат при наличии такой возможности или выбрать качественный генерик при невозможности назначить оригинальный препарат по финансовым соображениям.
Может ли практический врач в РФ заниматься оценкой качества генериков? Конечно нет, хотя многие врачи пытаются это делать. Однако это не входит в задачи практического врача, который просто должен иметь на рабочем столе источник достоверной информации о ЛС, в том числе о том, какой препарат является оригинальным, а какой генерическим, какой генерик относится к группе «А» и является качественным, а какой к группе «В» и не может быть заменой оригинальному ЛС и более качественному генерику. Тогда практический врач будет спокойно назначать то ЛС, которое считает нужным для данного больного. Кроме того, в этом случае врач должен иметь гарантию, что пациент в аптеке получит именно то ЛС (с тем торговым наименованием), которое выписано в рецепте. В нашей стране, к сожалению, нельзя допускать выписку рецептов по МНН, так как на фармацевтическом рынке присутствует огромное количество генерических ЛС очень различного качества. Врач же, как лицо юридически отвечающее за качество лечения пациента, должен быть уверен, что пациент получит именно то ЛС, которое он больному назначил.
Что делать пациенту в РФ? Начать задавать правильные вопросы врачу и государству, а также перестать экономить на здоровье. Хорошо известно, что население нашей страны на биологические добавки тратит в 10 раз больше средств, чем на лекарства [17].
Но, наверное, больше всего вопросов по поводу того, почему врач не имеет информации о том, какое торговое наименование соответствует оригинальному препарату, а какое генерическому. Почему в РФ отсутствует справочник ЛС, где это было бы указано? Ответственность за недостаточную информированность врача несет государство, которое должно обеспечивать все категории населения непредвзятой и доказательной информацией в отношении ЛС [18]. Полноценное лекарственное средство – это лекарственный продукт плюс полноценная информация о нем. Безусловно, наиболее простым решением проблемы с точки зрения практического врача был бы допуск на фармацевтический рынок РФ только качественных генерических ЛС.

Литература
1. Мешковский А.П. Место дженериков в лекарственном обеспечении. Фарматека. 2003; 3: 103–4.
2. Белоусов Ю.Б. Дженерики – мифы и реалии. Remedium. 2003;7–8: 4–9.
3. Иванов С. Выступление на встрече с членами Общественной палаты. Заседание, посвященное промышленности и инновациям. МГТУ имени Баумана. 2007; 18.05.
4. Панюшин Р. Оригинальные и дженериковые препараты: единство или борьба противопо¬ложностей? Фарм. вестник. 2003; 16: 23–25.
5. ЕМЕА, The rules governing medicinal products in the European Union Investigation of Bioavailability and Bioequivalence.1998; 3C: 231–244.
6. FDA, Electronic Orange Book. Approved Drug Products with Therapeutic Equivalence Evaluations, 23th Edition, 2003.
7. Материалы семинара «Совершенствование процесса регистрации и контроль качества лекарственных средств». Казахст. //Фармацевтический вестник. 16.10.2005.
8. Верткин А.Л., О.Б.Талибов О.Б. Генерики и эквивалентность – что стоит за терминами. Неотложная терапия. 2004; 1–2: 16–17.
9. Чумак В. Т. Оборот лекарственных средств в Украине. Проблемы и перспективы. Материалы І Международной конференции «Клинические испытания лекарственных средств в Украине». Киев, 2006, ноябрь.
10. Nightingale CH. A survey of the Quility of Generic Clarithromydn Product from 13 Countries. Clin Drug Invest 2000; 19: 293–05.
11. A UK–based retrospective study by CRAWFORD et al Seizure. 1996; 5: 1–5.
12. Насонова В.А. Особенности применения различных форм диклофенака натрия при болезнях мягких тканей плечевого пояса и боли в нижней части спины. Consilium medicum. 2006; 2 (8): 45–48.
13. Недогода С.В., Марченко И.В., Чаляби Т.А. Сравнительная антигипертензивная эффективность генериков ангиотензинпревращающего фермента эналаприла (ренитека, энапа, эднита, инворила, энваса и энама) и стоимость лечения у больных гипертонической болезнью. Артериальная гипертония. 2000; 1: 52–5.
14. Сущук Е.А. Симпозиум «Исследования по биоэквивалентности препаратов как основа для рационального использования генериков». Конгресс «Человек и лекарство», 3 апреля 2006 г.
15. The rules governing medicinal products in the European Union. Investigation of Bioavailability and Bioequivalence.1998; 3C: 231–244.
16. Марцевич С.Ю., Кутишенко Н.П., Дмитриева Н.А., Белолипецкая В.Г. Выбор лекарственного препарата в кардиологии: на что должен ориентироваться практический врач? Кардиоваскул. тер. и профилактика. 2004; 4: 77–82.
17. Гарматина Ю. Реклама против закона. //АиФ–Здоровье. 2004; 11, март: 2.
18. Рабочее совещание ВОЗ: Политика в области информации о ЛС в странах Европы и в Новых Независимых Государствах. Хиллеред. 1996; 27.03–2.04
Источник


