В ЕАЭС выстроена наднациональная модель регулирования общего рынка лекарств
О перспективах общего рынка лекарственных средств Евразийского экономического союза рассказал директор департамента технического регулирования и аккредитации Евразийской экономической комиссии Тимур Нурашев на научно-практической конференции «Экспертиза и регистрация лекарственных средств в ЕАЭС» – «РЕГЛЕК – ЕАЭС 2021».
Учитывая, что полный переход к единому рынку лекарств ЕАЭС завершится в 2025 году, особенно важной для производителей стала информация об изменении в 2020–2021 годах ряда переходных периодов.
В частности, в конце 2020 года для четырех стран ЕАЭС (Армении, Беларуси, Казахстана и Кыргызстана) для заявителей продлена возможность до 1 июля 2021 года подавать заявление на регистрацию лекарственного препарата по национальным правилам. Россия с 1 января 2021 года полностью перешла на регистрацию новых лекарственных препаратов по правилам ЕАЭС.
С 1 марта 2021 года вступила в силу Фармакопея ЕАЭС. С этой даты новые регистрационные досье, подаваемые по правилам ЕАЭС, должны содержать ссылки на Фармакопею ЕАЭС (при отсутствии необходимых статей в Фармакопее ЕАЭС указываются ссылки на фармакопеи, принятые в странах Союза).
Производители, которые успели подать документы до 1 марта 2021 без учета требований Фармакопеи ЕАЭС, должны перейти на нее до 1 января 2026 года, используя процедуру внесения изменений в регистрационное досье лекарственного препарата.
Для фарминспекторатов установлен единый предельный срок для выдачи национальных сертификатов GMP в государствах до 31 декабря 2021 года (для целей экспорта за пределы ЕАЭС выдача национальных сертификатов возможна до 31 декабря 2022 года).
Таким образом, с 2022 года все инспектораты ЕАЭС перейдут на союзные Правила надлежащей производственной практики при инспектировании производств лекарственных препаратов, предназначенных для общего рынка лекарственных средств в рамках ЕАЭС.
В 2021 году производителям лекарств предоставлена возможность при их регистрации по правилам ЕАЭС подавать имеющиеся национальные документы, подтверждающие соответствие требованиям GMP, до окончания срока их действия (фактически до 31 декабря 2024 года).
Директор департамента ЕЭК Тимур Нурашев отметил, что во избежание рисков создания пиковой нагрузки в работе регуляторов к концу переходного периода важно, чтобы переход к единому рынку лекарств ЕАЭС был плавным со стороны всех участников этого процесса.
Источник
Информация Коллегии ЕЭК от 03.01.2021 «Что изменится в регулировании доступа лекарственных препаратов на рынок ЕАЭС с 1 января 2021 года»
На правовом портале Евразийского экономического союза опубликовано решение Совета Евразийской экономической комиссии от 23 декабря о продлении на шесть месяцев (до 1 июля 2021 года) возможности для фармацевтических производителей выбирать регистрацию новых лекарств по национальной процедуре в четырех союзных государствах (Республике Армения, Республике Беларусь, Республике Казахстан и Кыргызской Республике).
Это означает, что с 1 января по 30 июня 2021 года в названных странах сохранится возможность регистрации лекарственных препаратов по национальным процедурам. 30 июня 2021 года — последний день, когда заявители смогут подать документы на регистрацию новых лекарственных препаратов в соответствии с законодательством государств ЕАЭС. Завершится один из основных переходных периодов общего рынка лекарственных средств для медицинского применения.
С 1 июля 2021 года (а в Российской Федерации — с 1 января 2021 года) новые лекарственные препараты (то есть лекарственные препараты, не имеющие действующих регистрационных удостоверений государств-членов Союза) могут быть зарегистрированы только в соответствии с Правилами регистрации и экспертизы лекарственных средств для медицинского применения, утвержденными Решением Совета ЕЭК N 78 от 3 ноября 2016 года.
До 31 декабря 2025 года для лекарственных препаратов, зарегистрированных в странах Союза, будут доступны процедуры внесения изменений и подтверждения регистрации, а также продление срока действия регистрационного удостоверения для срочных регистрационных удостоверений в соответствии с законодательством государств ЕАЭС.
Участники союзного рынка должны учитывать, что все регистрационные удостоверения, выданные по «национальным» правилам государств-членов, действительны до окончания срока их действия, но не позднее 31 декабря 2025 года.
Производителям, которые еще не сделали в связи с этим необходимых шагов, необходимо спланировать процесс внесения изменений в регистрационное досье для перехода к наднациональному регулированию, выбрать референтное государство, которое будет проводить экспертизу всех материалов досье и лабораторную оценку качества препарата, и уложиться с подачей документов по процедуре приведения в соответствие с требованиями международных договоров и актов, составляющих право Союза, в срок до 31 декабря 2025 года.
На общем рынке лекарственных средств ЕАЭС могут обращаться лекарственные препараты, качество, эффективность и безопасность которых оценена в соответствии с требованиями Союза, учитывающими наилучшие мировые практики. Это обеспечивается обязательным соблюдением надлежащих практик (GxP), подтверждается документами регистрационного досье, подготовленного в формате CTD, и полученными результатами лабораторной оценки качества образцов лекарственного препарата.
Источник
Компас фармрынка


Наряду с нормативными правовыми актами ЕАЭС она призвана служить фактором развития конкурентоспособной и экспортно ориентированной фарминдустрии государств — членов ЕАЭС. Она является основополагающим документом, необходимым для регистрации лекарственных средств в рамках ЕАЭС, устанавливает критерии доступа лекарственных средств по качеству на единый рынок.

Основные принципы и подходы к созданию Фармакопеи ЕАЭС сформулированы в Концепции гармонизации фармакопей государств-членов ЕАЭС, утвержденной решением Коллегии ЕЭК от 22 сентября 2015 года. Для ее создания из представителей государств — членов ЕАЭС был сформирован Фармакопейный комитет ЕАЭС, который начал свою деятельность с февраля 2017 года. За прошедшее время были заложены методологические основы разработки общих и частных фармакопейных статей, определена структура Фармакопеи.
Первая часть первого тома Фармакопеи Союза включает 157 гармонизированных общих фармакопейных статей (монографий), содержащих общие сведения о применении фармакопейного анализа и его методиках, методах биологических и микробиологических испытаний, реактивах, приборах и аппаратах для анализа качества как уже обращающихся на рынке, так и новых, еще только разрабатываемых лекарств.
В планах дальнейшей работы — наполнение документа новыми общими и частными статьями с подготовкой издания следующих ее томов.
Разрабатываются общие фармакопейные статьи, предусмотренные для включения во вторую часть первого тома Фармакопеи ЕАЭС.
В условиях глобализации мирового фармрынка основным вектором в развитии современных фармакопейных стандартов является их гармонизация. Гармонизация Фармакопеи ЕАЭС необходима для интегрирования ее в глобальную систему регулирования обращения лекарственных средств.
В соответствии с Концепцией гармонизация Фармакопеи ЕАЭС осуществляется на основе национальных фармакопей государств — членов ЕАЭС (Республики Беларусь, Республики Казахстан и Российской Федерации) и основных фармакопей мира.
В качестве основных фармакопей мира приняты Европейская, Британская и Фармакопея США, при этом Европейская определена как базовая среди них. В процессе создания Фармакопеи ЕАЭС ведущая роль отведена руководству Всемирной организации здравоохранения «Надлежащая фармакопейная практика (GPhP)».
Помимо фармакопейных стандартов в ряде случаев в Фармакопее ЕАЭС использованы правила и руководства ведущих международных организаций, например Международного совета по гармонизации технических требований к лекарственным препаратам для медицинского применения (ICH), ВОЗ.
Наряду с гармонизированными фармакопейными статьями Фармакопея ЕАЭС содержит значительное число фармакопейных статей, испытаний и (или) методик их проведения, не имеющих аналогов в других фармакопеях.
Фармакопея ЕАЭС представляет собой свод региональных требований и положений, устанавливающих необходимый уровень качества лекарственных средств на фармацевтическом рынке ЕАЭС. Требования Фармакопеи ЕАЭС распространяются на лекарственные средства как для медицинского, так и для ветеринарного применения.
Фармакопея ЕАЭС занимает центральное место в системе стандартизации лекарственных средств. Она определяет уровень качества, устанавливаемый спецификациями производителей лекарственных средств, который должен быть не ниже фармакопейных требований. На основании спецификации производителя по согласованию с уполномоченными органами государств — членов Союза составляется нормативный документ по качеству лекарственного препарата, предназначенный для контроля его качества в пострегистрационный период его обращения на фармрынке ЕАЭС.

Фармакопея Союза неразрывно связана с нормативными правовыми актами ЕАЭС в области обеспечения качества лекарственных средств, в частности с правилами надлежащих фармацевтических практик (GхP). Они призваны обеспечивать соответствие требованиям Фармакопеи от серии к серии лекарственного средства, для каждой единицы лекарственной формы. С другой стороны, необходимость выполнения требований Фармакопеи стимулирует постоянное внедрение и поддержание правил GхP в сфере обращения лекарственных средств.
Фармакопея ЕАЭС имеет статус региональной. Ее требования являются обязательными для всех предприятий и организаций государств-членов Союза, занимающихся производством, изготовлением, реализацией, хранением, экспертизой, регистрацией, контролем качества и применением лекарственных средств.
Утверждение и введение в действие Фармакопеи ЕАЭС обязывает заявителей (производителей), зарегистрировавших лекарственные препараты по правилам ЕАЭС, внести изменения в регистрационные досье на лекарственные препараты до 31 декабря 2025 года. Все вновь подаваемые заявления на регистрацию с 1 марта 2021 года должны содержать в спецификациях модуля 3 регистрационного досье и в нормативном документе по качеству ссылки на Фармакопею ЕАЭС. В связи с утверждением первой части первого тома Фармакопеи ЕАЭС эти изменения касаются общих требований к испытаниям, методам их проведения и используемым реактивам.
Процедура утверждения и введения в действие фармакопейных требований не происходит одномоментно. Период времени между утверждением и введением в действие фармакопейных требований необходим заявителям (производителям) для подготовки к предстоящим изменениям, которые связаны с оценкой внутренних ресурсов и возможностей, перестройкой и обновлением отдельных процессов и процедур, модификацией существующих методов и (или) методик испытаний, их валидацией или верификацией и т.п.
Введение в действие Фармакопеи ЕАЭС предусмотрено спустя полгода после ее официального утверждения, т.е. с 1 марта 2021 года. Региональный статус, а также то, что фармакопейные статьи создаются впервые, обосновывают ответственность и высокие требования к деятельности Фармакопейного комитета ЕАЭС. Его задачи на ближайшую перспективу включают:
— разработку и выпуск последующих частей (2-4 части) первого тома Фармакопеи ЕАЭС, содержащих общие фармакопейные статьи;
— развитие методологических основ создания Фармакопеи ЕАЭС в виде руководств по разработке частных фармакопейных статей Фармакопеи ЕАЭС на различные лекарственные средства (субстанции для фармацевтического применения химического происхождения, радиофармацевтические лекарственные препараты, растительные лекарственные средства, биологические лекарственные средства и др.);
— развитие организационной структуры ФК ЕАЭС в виде специализированных экспертных групп различного профиля и обеспечение их успешного функционирования.

Дальнейшая деятельность ФК ЕАЭС будет связана с разработкой и выпуском второго тома Фармакопеи ЕАЭС, включающего частные фармакопейные статьи на субстанции для фармацевтического применения и лекарственные препараты. Это потребует создания собственных стандартных образцов, используемых для испытаний лекарственных средств наряду со стандартными образцами фармакопей, с которыми гармонизирована Фармакопея ЕАЭС. Задачей перспективного развития Фармакопеи ЕАЭС является создание экспериментальной базы в виде региональной сети лабораторий, аккредитованных на международном уровне.
Наконец, создание и интегрирование Фармакопеи ЕАЭС в глобальную систему регулирования лекарственных средств должно осуществляться на основе принципа транспарентности с широким вовлечением национальных регуляторных органов и профессионального сообщества в обсуждение фармакопейных стандартов на этапах разработки и правоприменения.
Ардак Уринбасаровна Тулегенова руководит центром по разработке и совершенствованию Государственной фармакопеи Республики Казахстан и Фармакопеи ЕАЭС Национального центра экспертизы лекарственных средств и медицинских изделий Минздрава Республики Казахстан. Доктор фармацевтических наук, профессор, почетный член Национальной академии наук Республики Казахстан.
Источник
Лекарства вступают в союз

Общие правила обращения лекарственных препаратов и медицинских изделий для членов Евразийского экономического союза (ЕАЭС) — России, Армении, Белоруссии, Казахстана и Киргизии — начали действовать с мая 2017 года. Объединение рынков будет идти поэтапно. И к 2025 году на территории союзных государств правила производства и реализации фармацевтических препаратов и медизделий должны быть полностью унифицированы.
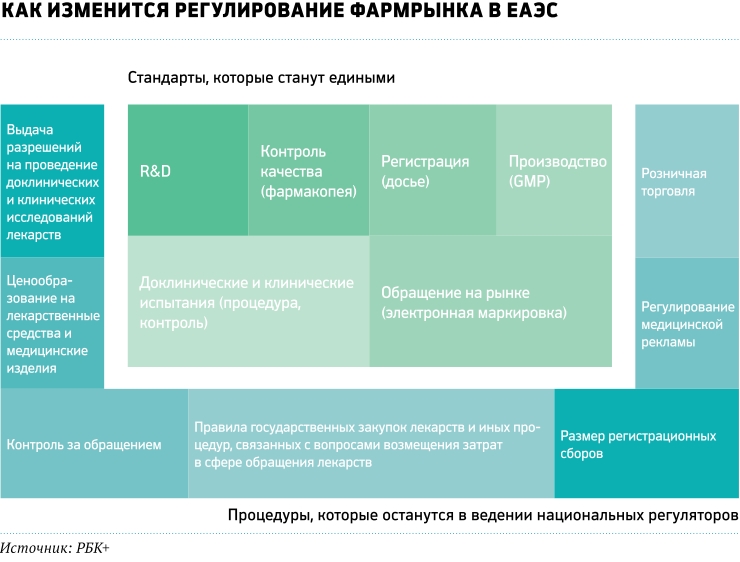
Как в Европе
Пакет из 26 документов, включающий решения Совета Евразийской экономической комиссии (ЕЭК) и рекомендацию Коллегии ЕЭК, должен сделать фармацевтический рынок пяти стран, подписавших эти документы, похожим на европейский. Лекарства и медицинские изделия будут производиться и реализовываться по единым правилам. О том, что за основу взята европейская модель общего регуляторного пространства, говорили еще на стадии согласования новых принципов. В феврале этого года министр по техническому регулированию ЕЭК Валерий Корешков на встрече в Страсбурге с руководством Европейского директората по качеству лекарственных средств и здравоохранению (EDQM) рассказал о создании единой фармакопеи (свода стандартов, устанавливающих норму качества сырья для производства медицинских препаратов) в странах ЕАЭС. В перспективе принятие документа позволит унифицировать евразийские и европейские правила. «Мы ставим задачу добиться в ЕАЭС как минимум такой же свободы обращения качественных лекарств (как в ЕС. — РБК+), отвечающих потребностям здравоохранения и международной торговли», — заявил Валерий Корешков в ходе встречи в Страсбурге.
Все разработанные и вступившие в силу в мае нормативные акты — это документы прямого действия, то есть на территории всех пяти государств они не требуют принятия дополнительных законов для ратификации. По замыслу разработчиков, общее экономическое пространство снизит бюрократическое давление на фармацевтическую отрасль и упростит доступ качественной и современной продукции на рынок всех стран-участниц. Производителям обещано значительное снижение административных издержек, а пациентам — снижение цен на все основные лекарства и повышение их качества.
Оцифровка лекарств
С 1 января 2019 года будет введена единая электронная маркировка лекарств. Сейчас электронная маркировка введена только в Армении и Белоруссии, и у каждой страны она своя. Поэтому за ближайший год предстоит разработать единую систему в формате DataMatrix (двумерный матричный штрих-код) и создать так называемые шлюзы в национальных информационных системах для подключения к единой базе данных лекарственных препаратов экономического союза. Технологическая совместимость — одно из основных требований договора ЕАЭС.
Новая маркировка сделает прозрачным происхождение той или иной партии лекарств или медизделий, гарантировав их качество конечному потребителю, и параллельный экспорт, говорит исполнительный директор Союза профессиональных фарморганизаций (СПФО) Лилия Титова. Речь идет о практике ввоза в страну продукции без ведома производителя, что абсолютно законно, например, в Армении, но не допускается в других странах экономического союза. Часто источником таких поставок оказываются гуманитарная помощь или лекарства, закупленные международными организациями. Введение новой электронной маркировки не позволит совершать такие маневры незаметно.
До 31 декабря 2020 года производитель лекарств имеет право выбирать, по каким правилам регистрировать препараты — национальным или общим. Правда, потом зарегистрированные по старинке лекарства все равно придется перерегистрировать — 31 декабря 2025 года регистрационные свидетельства старого образца перестанут действовать. Еще одна отсрочка действует в отношении подтверждающих документов: до 31 декабря 2018 года при подаче досье на регистрацию производитель может показать не сертификат общего для ЕАЭС стандарта качества производства GMP, а местный документ, подтверждающий соответствие производственной площадки национальным требованиям по качеству. Предполагается, что вся процедура с момента подачи досье лекарственного препарата на регистрацию до выпуска его в обращение займет от семи до десяти месяцев.
«Единая для всех стран процедура регистрации пока не запущена, так как местные власти должны постановлением правительства утвердить размер госпошлин», — пояснила РБК+ Лилия Титова.
В России такой нормативный акт был принят еще в марте 2017 года. В Налоговый кодекс России внесены соответствующие изменения. Так, за проведение экспертизы лекарственного препарата для медицинского применения при регистрации по новым правилам производитель должен заплатить государству 325 тыс. руб., за подтверждение регистрации — 145 тыс., за приведение регистрационного досье в соответствие с требованиями ЕАЭС — 75 тыс. руб. Причем уплата пошлин в одной стране не освобождает от уплаты пошлин в другой.
Лечение партнеров
Основной проблемой для формирования единого фармацевтического пространства в пяти странах союза остается «двухслойная» структура регулирования, считают эксперты. Ведь кроме свода общих союзных правил остаются внутренние, а они зачастую противоречат друг другу от страны к стране. Например, неразрешенной проблемой остается правило «третий лишний» при бюджетных закупках лекарственных препаратов, работающее в России. Эта мера призвана повысить шансы отечественных производителей на участие в государственном заказе. В контексте открытого рынка она может сменить знак на противоположный, открыв огромный российский рынок госзакупок не самым известным фармпроизводителям из дружественных государств с более низкими ценами, чем у отечественных производителей.
«Правила взаимозаменяемости препаратов, которые разработаны в России лучше, чем в других странах, и правило «третий лишний» делают наш рынок более открытым для лекарств из других стран ЕАЭС, чем их рынки открыты для нас», — считает директор Института экономики здравоохранения НИУ ВШЭ Лариса Попович.
Система регулирования во всех странах ЕАЭС сильно отличается от российской, поэтому нашей стране придется значительно сильнее подстраиваться к единому регулированию, чем другим участникам соглашения. Например, никто не собирается отдавать предпочтение российским лекарствам в госзакупках, так как эти правила по-прежнему остаются на национальном уровне. Все это кажется экспертам несправедливым, учитывая, что Россия — единственный крупный фармрынок в регионе, где уровень потребления лекарств на порядок выше, чем в других странах союза.
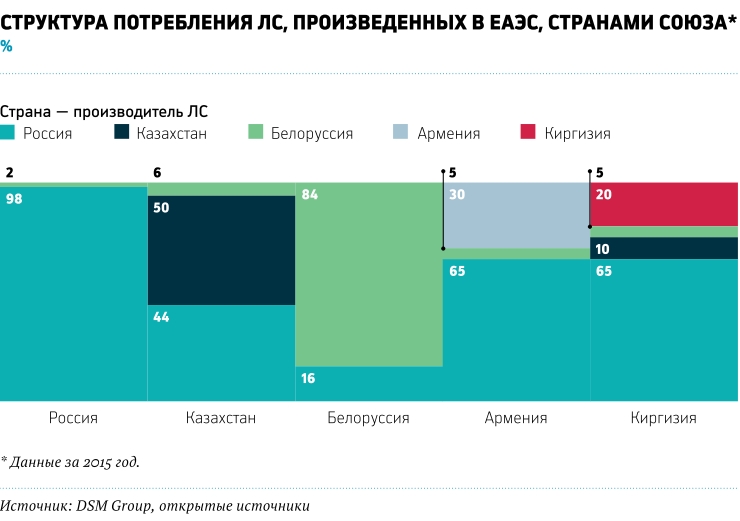
Общий объем фармрынка ЕАЭС, по данным DSM Group, в 2015 году превысил $20,6 млрд. Доля России достигает 85% и составляет около $17,4 млрд, Казахстана — 9%, или порядка $1,8 млрд, Белоруссии — 5%, или чуть более $1 млрд. Объемы фармрынков Армении и Киргизии составляют 0,5 и 1% соответственно от общего объема рынка (см. график).
«Для российского потребителя лекарств и производителей ждать положительных изменений в связи с объединением вряд ли приходится, — говорит Лариса Попович. — Родовые пятна в регулировании остаются, а количество обязательств увеличивается. Я бы не стала надеяться и на снижение цен для конечного потребителя. Вспомните, что правило «третий лишний» привело к росту цен, так как отбор осуществляется не по цене, а по стране происхождения». По мнению эксперта, Россия идет хорошо известным путем помощи странам из «своего» лагеря. Поэтому от объединения стоит ожидать скорее политический эффект, чем экономический.
Опыт ЕС
Единый рынок лекарственных средств (ЛС) в рамках Европейского экономического сообщества (ЕЭС) был создан в 1992 году. Сегодня он объединяет 27 стран. Общие правила его функционирования сначала принимались на уровне ЕЭС, а затем Европейского союза (ЕС). Постепенно они вытесняли национальное законодательство. На данный момент право ЕС проникло в регулирование почти всех стадий производства и продажи фармацевтических средств.
С 1995 года на территории ЕС действует единая система допуска ЛС на рынок, так называемая централизованная процедура. Однако государства сохранили в определенных случаях собственные национальные процедуры. Преимущество централизованной регистрации — действие разрешения во всех странах Евросоюза, если заключение относительно рассматриваемого нового лекарства дает специально созданное Европейское агентство по оценке лекарственных препаратов (European Medicines Evaluation Agency, EMEA). На основании этого заключения Комитет по патентованным лекарственным препаратам (Committee on Proprietary Medicinal Products, CPMP) Евросоюза принимает решение о допуске лекарства на рынок.
Однако единый порядок применим лишь в отношении определенного и очень ограниченного круга препаратов: являющихся в значительной степени инновационными, созданными в результате определенных биотехнологических процессов, обладающих новыми видами терапии и содержащих новые действующие вещества. Национальная процедура по допуску лекарств на внутренний рынок осуществляется на основе законодательства отдельной страны, в которой также отражены требования директив ЕС в фармацевтической сфере. При этом существует возможность упрощенной регистрации препаратов, разрешение на которую уже было получено в одной из стран ЕС.
По мнению авторов исследования Санкт-Петербургского филиала НИУ ВШЭ «Влияние права Европейского союза на регулирование фармацевтических средств государств-членов» (2014), внутренний рынок лекарственных препаратов в ЕС так и не был создан — для производителей и импортеров некоторых видов ЛС по-прежнему существует необходимость получать до 27 разрешений допуска на фармацевтический рынок. «Сложившаяся система может быть оптимизирована путем введения принципа европаспорта (Europass), который успешно применяется в других областях», — говорится в исследовании.
Источник


