- Изменился перечень орфанных заболеваний, препараты для лечения которых закупаются на федеральном уровне
- Орфанные заболевания
- Препаратный маневр
- Как пациенты получают лекарства
- В каких регионах недостаточно тратят на лекарства
- Причины проблем с диагностикой орфанных заболеваний
- Поможет ли централизация закупок решению проблемы
Изменился перечень орфанных заболеваний, препараты для лечения которых закупаются на федеральном уровне
 |
| bogdan.hoda / Depositphotos.com |
На федеральный уровень передано финансирование лекарственного обеспечения граждан, страдающих неуточненной апластической анемией, наследственным дефицитом факторов II (фибриногена), VII (лабильного), X (Стюарта-Прауэра). В связи с этим Правительство РФ уточнило (Постановление Правительства РФ от 5 июня 2020 г. № 829):
- перечень жизнеугрожающих и хронических прогрессирующих редких (орфанных) заболеваний, приводящих к сокращению продолжительности жизни граждан или их инвалидности;
- правила формирования перечней лекарств и минимального ассортимента препаратов, необходимых для оказания медпомощи;
- порядок функционирования единой государственной информационной системы в сфере здравоохранения;
- программу госгарантий бесплатного оказания гражданам медпомощи на 2020 г. и плановый период 2021 и 2022 гг.
Все важные документы и новости о коронавирусе COVID-19 – в ежедневной рассылке Подписаться
Источник
Орфанные заболевания
Один из миллиона, особенный, уникальный –
эти эпитеты подойдут гениям и тем, кому повезло меньше, –
людям с редкими болезнями.
Им не помогают обычные лекарства, они редко доживают до старости
и часто выглядят не так, как другие люди.
Мы уже неоднократно обращались к теме орфанных заболеваний. Актуальность обсуждения данной проблемы обусловлена повышенным вниманием к ней не только в России, но и во всём мире. Связано это в основном с высоким уровнем экономического и социального бремени редких болезней.
Напомним, что орфанными считаются заболевания, затрагивающие небольшую часть популяции. Не существует единого, широко принимаемого определения редких заболеваний. Некоторые определения полагаются на количество людей, живущих с заболеванием, другие могут включать иные факторы, например, доступность лечения болезни или возможность облегчения ее течения. В США Акт о редких заболеваниях (Rare Disease Act) 2002 года определяет редкие болезни как «болезни или состояния, затрагивающие менее 200 000 людей в США» («any disease or condition that affects less than 200,000 persons in the United States»), или примерно 1 человека из 1500. Низкий уровень в популяции обычно соответствует менее чем 1 из 2000. Болезни, не являющиеся угрожающими жизни, серьёзными хроническими, либо адекватно излечимыми, исключаются из определения. В России редкими предлагается считать заболевания с «распространенностью не более 10 случаев на 100 000 человек». В настоящее время в нашей стране Постановлением Правительства Российской Федерации от 26.04.2012 г. № 403 утвержден перечень из 24 редких (орфанных) заболеваний.
Таблица № 1
Перечень
жизнеугрожающих и хронических прогрессирующих редких (орфанных) заболеваний, приводящих к сокращению
продолжительности жизни граждан или их инвалидности
(утв. постановлением Правительства РФ
от 26 апреля 2012 г. № 403)
Заболевание
Код
заболевания
Пароксизмальная ночная гемоглобинурия (Маркиафавы-Микели)
Апластическая анемия неуточненная
Наследственный дефицит факторов II (фибриногена), VII (лабильного), X (Стюарта-Прауэра)
Идиопатическая тромбоцитопеническая
пурпура (синдром Эванса)
Дефект в системе комплемента
Преждевременная половая зрелость
центрального происхождения
Нарушения обмена ароматических
аминокислот (классическая фенилкетонурия, другие виды гиперфенилаланинемии)
Болезнь «кленового сиропа»
Другие виды нарушений обмена аминокислот
с разветвленной цепью (изовалериановая
ацидемия, метилмалоновая ацидемия,
пропионовая ацидемия)
Нарушения обмена жирных кислот
Другие сфинголипидозы:
болезнь Фабри (Фабри-Андерсона),
Нимана-Пика
Мукополисахаридоз, тип I
Мукополисахаридоз, тип II
Мукополисахаридоз, тип VI
Острая перемежающая (печеночная) порфирия
Нарушения обмена меди (болезнь Вильсона)
Легочная (артериальная) гипертензия
(идиопатическая) (первичная)
Юношеский артрит с системным началом
Примерно половина орфанных заболеваний обусловлена генетическими отклонениями. Симптомы могут быть очевидны с рождения или проявляться в детском возрасте. Проблема редких заболеваний особенно актуальна для педиатрии, так как 2/3 редких болезней манифестируют в раннем возрасте. Реже встречаются токсические, инфекционные или аутоиммунные «сиротские» болезни. Причинами их развития могут быть наследственность, ослабление, плохая экология, высокий радиационный фон, у мамы и у самих детей в раннем возрасте. Большинство орфанных заболеваний – хронические. Они в значительной мере ухудшают качество жизни человека и могут стать причиной летального исхода. Для большинства таких болезней не существует эффективного лечения. Основа терапии таких больных – улучшение качества и увеличение продолжительности жизни пациентов.
Впервые на территории Российской Федерации орфанные заболевания получили юридический статус в 2011 году с принятием 21 ноября2011 г. Федерального закона № 323-ФЗ «Об основах охраны здоровья граждан в Российской Федерации». В соответствии с ч. 9 ст. 83 данного федерального закона граждане РФ, страдающие жизнеугрожающими и хроническими прогрессирующими редкими (орфанными) заболеваниями, имеют право на обеспечение препаратами, предназначенными для лечения этих заболеваний, за счет средств субъектов РФ. Однако большинство региональных бюджетов орфанной нагрузки не выдерживают и даже ограниченный перечень из 24 редких заболеваний обслуживают с трудом [3].
На сегодняшний день на территории Брянской области проживают 107 пациентов, страдающих редкими (орфанными) заболеваниями, из них 55 детей. Для обеспечения указанной категории пациентов необходимо 300 миллионов рублей в год, дефицит финансирования программы в регионе составляет 87%.
Таблица № 2
Количество пациентов Брянской области,
страдающих редкими (орфанными) заболеваниями
Заболевание
Численность
Апластическая анемия неуточненная (D61.9)
Гемолитико-уремический синдром (D59.3)
Дефект в системе комплемента (D84.1)
Идиопатическая тромбоцитопеническая пурпура (синдром Эванса) (D69.3)
Мукополисахаридоз, тип VI (E 76.2)
Нарушения обмена ароматических аминокислот (классическая фенилкетонурия, другие виды
гиперфенилаланинемии) (Е70.0, Е70.1)
Нарушения обмена меди (болезнь Вильсона)
(E 83.0)
Наследственный дефицит факторов
II (фибриногена), VII (лабильного),
Х (Стюарта-Прауэра) (D68.2)
Незавершенный остеогенез (Q78.0)
Пароксизмальная ночная гемоглобинурия
(Маркиафавы-Микели) (D59.5)
Юношеский артрит с системным началом
(M 08.2)
Стоимость лечения пациентов с различными редкими заболеванием различная. Например, для лечения галактоземии необходимо специальное лечебное питание: без галактозы и лактозы. Стоимость его лишь незначительно отличается от стоимости обычного питания для детей. Но лечение других пациентов может составлять сотни тысяч в месяц. При составлении Перечня не учитывалась стоимость лечения и с общечеловеческой точки зрения, включение заболеваний в перечень вне зависимости от стоимости лекарства подчёркивает равные права больных в доступности современного лечения.
Депутаты государственной Думы, органы исполнительной власти субъектов РФ и общественные пациентские организации неоднократно поднимали вопрос о необходимости привлечения средств федерального бюджета к софинансированию региональных программ обеспечения лекарственными препаратами лиц, страдающих редкими (орфанными) заболеваниями. Проблемы финансирования программ в регионах усугубляется отсутствием единой модели оказания медицинской помощи, в том числе лекарственными препаратами, данной категории граждан. Каждый субъект РФ решает эти проблемы по-своему. При этом есть субъекты, в которых ориентированность на сохранение жизни пациента взята за основу при принятии решений по лечению больных с редкими заболеваниями (республики Башкоркостан, Дагестан, Коми, Калмыкия, Московская область и другие). В других же (республика Татарстан, Нижегородская область) добиться обеспечения лекарственными препаратами возможно только через судебные решения, которые также не сразу исполняются региональными властями.
Еще около 30 лет назад препаратов, предназначенных для лечения орфанных заболеваний, практически не было. Из-за узости рынка большинство фармкомпаний разработку и лонч таких лекарственных средств (ЛС) считали непривлекательными. По версии Tufts Center, чтобы вывести орфанный препарат на рынок США, компания должна потратить не менее 1,2 млрд дол. Ограниченное число пациентов для проведения клинических исследований лишь усугубляло ситуацию. В 1983 году в США был издан Orphan Drug Act, в котором впервые появилось понятие «орфанные заболевания». В документе прописывались преференции для производителей орфанных препаратов, в том числе налоговые льготы в размере 50% на проведение клинических испытаний на территории США, упрощение процедуры лицензирования и сокращение стоимости регистрации, предоставление эксклюзивных прав на продажу сроком на семь лет.
В 90-е годы аналогичные документы были изданы в Сингапуре, Японии и Австралии, в 2000 году – в Европе.
Благодаря этим мерам количество орфанных ЛС возросло. По прогнозам EvaluatePharma, мировой объем продаж препаратов для лечения редких болезней с 90 млрд дол., зафиксированных в 2013 году, через пять лет подберется к отметке в 127 млрд дол. Среднегодовой темп роста сегмента составит 7,4%, что вдвое выше аналогичного показателя для Rx-препаратов.
На сегодняшний день самой дорогостоящей нозологией среди входящих в Перечень, является пароксизмальная ночная гемоглобинурия. Пароксизмальная ночная гемоглобинурия (ПНГ) – редко встречающееся приобретенное заболевание, вызванное нарушением эритроцитарной мембраны и характеризующееся хронической гемолитической анемией, перемежающейся или постоянной гемоглобинурией и гемосидеринурией, явлениями тромбоза и гипоплазией костного мозга. Это заболевание обычно впервые диагностируется у лиц в возрастной группе 20–40 лет, но может встречаться и у пожилых. При классической форме гемолиз (разрушение эритроцитов крови с выделением в окружающую среду гемоглобина) происходит в то время, когда больной спит (ночная гемоглобинурия), что может быть обусловлено небольшим снижением ночью рН крови. Однако гемоглобинурия (появление гемоглобина в моче.) наблюдается только примерно у 25% больных, причем у многих не в ночное время. Препарат, использующийся для лечения данного заболевания, – самый дорогостоящий в мире. Годовой курс лечения препаратом Солирис Soliris (МНН: экулизумаб) от швейцарской компании Alexion Pharma оценивается в 400 тыс. дол.
Elaprase (идурсульфаза, «Shire Human Genetic Therapies, Inc.») компенсирует недостаток одного из лизосомальных ферментов, генетически обусловленная нехватка которого развивается при синдроме Хантера (мукополисахаридозе II типа). Синдром Хантера – одна из форм мукополисахаридоза, возникает в результате дефицита ряда ферментов, что приводит к накоплению белково-углеводных комплексов и жиров в клетках. Болезнь проявляется в раннем возрасте (2–4 года) утолщением ноздрей, губ, языка,
тугоподвижностью суставов, задержкой роста. До двух лет отмечают такие признаки, как шумное дыхание (обструкция верхних дыхательных путей), риниты, паховые и пупочные грыжи. До появления Elaprase лечение синдрома Хантера ограничивалось паллиативной терапией. В России препарат зарегистрирован в 2008 году, стоимость лечения составляет 350 тыс. дол./год.
Naglazyme (гальсульфаза, «BioMarin Pharmaceuticals») – первый и единственный препарат для лечения синдрома Марото–Лами (мукополисахаридоза VI типа). Naglazyme замещает недостающий при этом заболевании фермент (арилсульфатаза). Заболевание характеризуется отложением дерматансульфата в различных тканях. Манифестирует к 2–3 годам жизни отставанием в росте, огрублением черт лица, напоминающим фенотип синдрома Гурлер, помутнением роговицы, деформациями скелета (поясничный кифоз, вальгусное искривление голеней), гепатоспленомегалией, тугоподвижностью суставов, грыжами. Умственная отсталость не характерна. Пациенты, принимающие Наглазим, смогли преодолеть больше ступенек лестницы и ходить на более далекие расстояния, чем раньше. Синдром Марото–Лами отмечен в настоящее время примерно у 1100 людей в мире. Стоимость лечения составляет около 350 тыс. дол./год.
Ещё один дорогостоящий препарат – Myozyme (альглюкозидаза альфа, «Genzyme Corporation») показан для лечения пациентов с болезнью Помпе (дефицитом альфа-глюкозидазы, которая необходима, чтобы разрушать гликоген – вещество, которое является источником энергии для организма). Симптомы болезни Помпе возникают из-за аномального накопления гликогена в клетках. В настоящее время болезнь Помпе не входит в Перечень, однако пациентские сообщества и медицинские организации ведут активную работу по расширению Перечня-24, в частности по включению в него болезни Помпе. Без заместительной терапии дети с ранним началом заболевания умирали в течение первого года жизни вследствие недостаточности кровообращения или инфекций респираторного тракта. Согласно результатам клинических испытаний Myozyme увеличивал период выживаемости без искусственной вентиляции легких у таких пациентов. Лечение ребенка этим препаратом в течение года стоит около 100 тыс. дол., взрослого – около 300 тыс. дол.
Aldurazyme (ларонидаза, «Genzyme Corporation») предназначен для ферментозаместительной терапии при мукополисахаридозе I типа (синдром Гурлера) – тяжелом прогрессирующем, часто жизнеугрожающем, заболевании. Стоимость лечения препаратом составляет 200 тыс. дол./год [5].
Улучшить ситуацию с обеспечением лекарствами пациентов, страдающих редкими заболеваниями, должны готовящиеся в настоящее время федеральные нормативно-правовые акты, в частности поправки в закон «Об обращении лекарственных средств» №61-ФЗ: изменения, дающие преференции орфанным препаратам, подготовлены, но пока не приняты. Также решения требует принципиальный вопрос – передача финансирования помощи гражданам, страдающим редкими заболеваниями, с регионального на федеральный уровень.
С 2008 года последний день февраля объявлен Днём редких заболеваний.
Источник
Препаратный маневр

Государственные расходы на закупку лекарственных препаратов для лечения различных орфанных (редких) заболеваний распределены среди российских регионов со значительным неравенством — на десять из них приходится 50% совокупных расходов всех субъектов. Об этом говорится в аналитическом отчете Центра стратегических разработок (ЦСР), с которым ознакомился РБК. Документ посвящен анализу возможностей централизации закупок лекарств для пациентов с редкими заболеваниями за счет средств федерального бюджета.
Совет ЦСР, созданного в 1999 году для анализа путей социально-экономического развития России, с июня 2020 года возглавляет глава Минэкономразвития Максим Решетников.
Как пациенты получают лекарства
В России к орфанным отнесено 260 заболеваний. Однако на текущий момент терапия только 28 из них финансируется государством: 11 — по федеральной программе «14 высокозатратных нозологий» и 17 — из региональных бюджетов по перечню 17 редких угрожающих жизни заболеваний. Как указывают авторы исследования ЦСР, по этой группе пациентов ведется централизованный учет, тогда как по остальным редким заболеваниям из списка, которых более 200, — нет.
По версии экспертов ЦСР, поэтапная централизация закупок лекарств за счет средств федерального бюджета (федерализация), а также создание запаса препаратов способны исключить или минимизировать существующие недочеты системы лекарственного обеспечения.
В каких регионах недостаточно тратят на лекарства
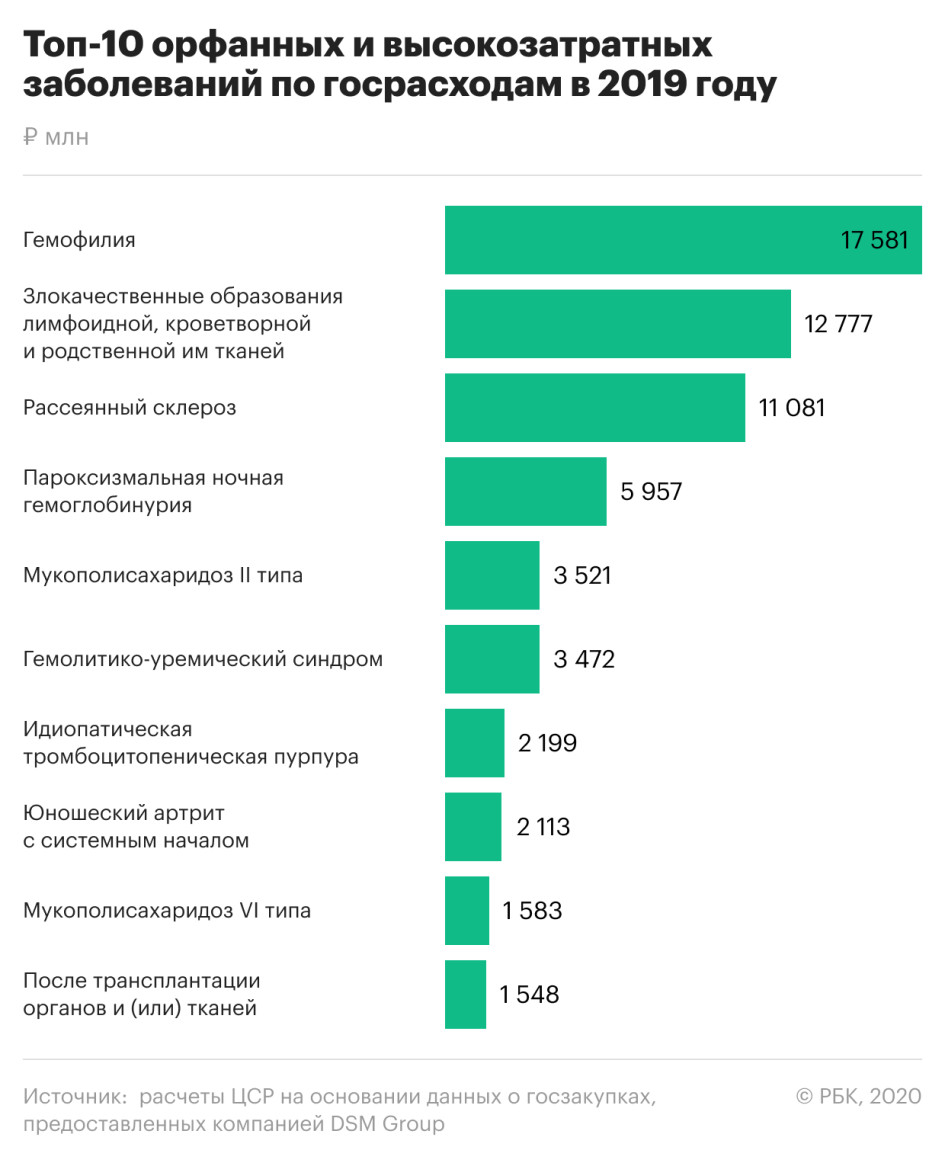
Три наиболее затратных для госбюджета орфанных заболевания — гемофилия (в 2019 году на закупку препаратов пошло более 17 млрд руб.), пароксизмальная ночная гемоглобинурия (почти 6 млрд руб.) и мукополисахаридоз II типа (более 3,5 млрд руб.). Как указывают авторы, на обеспечение пациентов этих нозологий лекарствами приходится более половины (56%) государственных расходов федерального и региональных уровней.
Федеральный бюджет на закупку лекарств для лечения редких заболеваний к июлю 2020 года был исполнен на 91%, говорится в докладе. Что касается расходов регионов на закупку таких препаратов, то здесь эксперты отмечают существенный дисбаланс — на десять регионов приходится 50% совокупных расходов всех субъектов. При этом доля Москвы свыше 17%, среди лидеров также Санкт-Петербург, Свердловская область, Краснодарский край, Челябинская область.
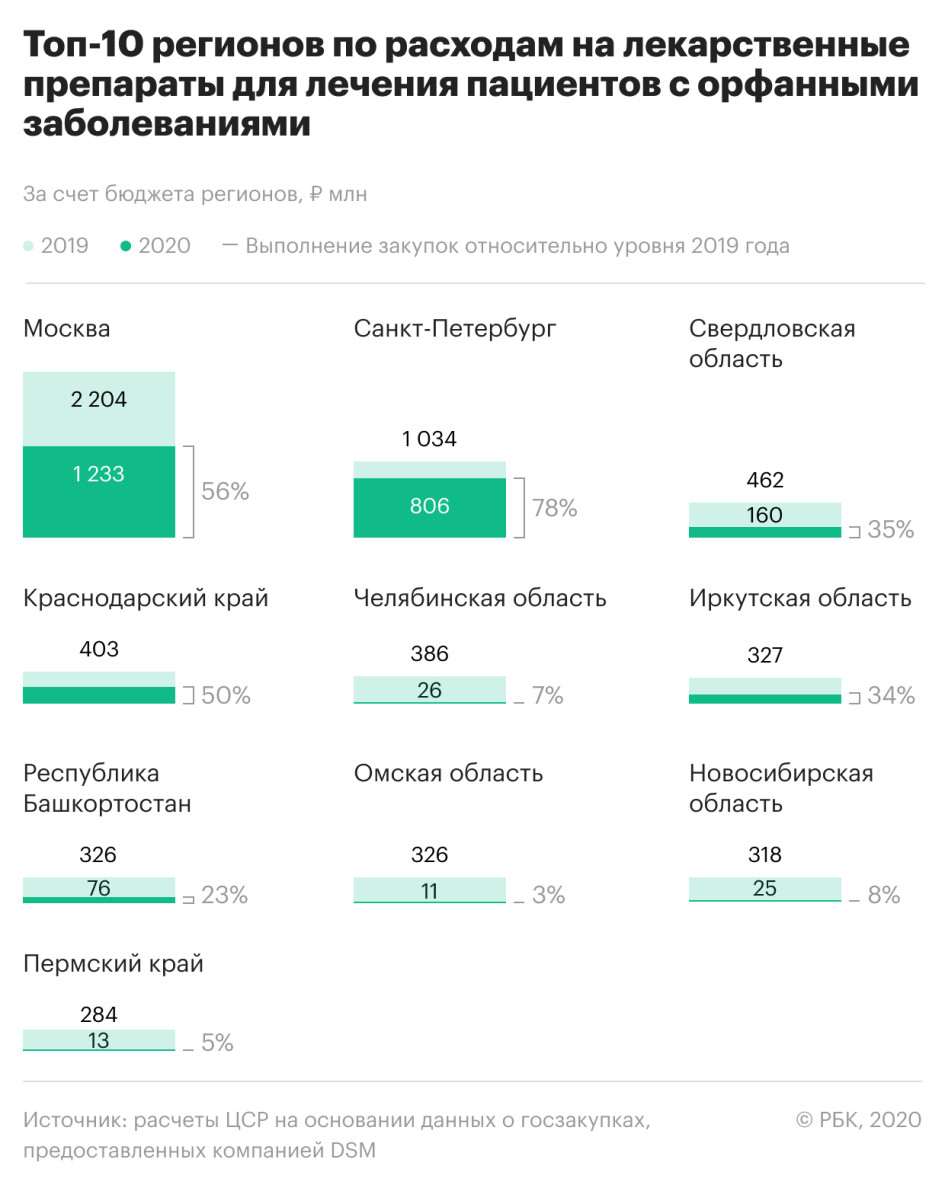
Среди регионов с наименьшим объемом выделяемых средств — Марий Эл, Крым, Пензенская, Владимирская, Брянская области. Средний объем финансирования, безусловно, зависит от состава пациентов с тем или иным заболеванием, поэтому он варьируется от региона к региону, уточняется в докладе, но при анализе расходов видно, что в наиболее проблемных регионах обеспеченность значительно ниже среднего.
В целом в большинстве регионов не хватает средств для обеспечения пациентов необходимыми лекарствами — по данным ежегодного бюллетеня экспертного совета по орфанным заболеваниям комитета Госдумы по охране здоровья, совокупный дефицит региональных бюджетов в 2019 году составлял 19%. В 2020 году, как ожидается, этот показатель будет равен 14%.
РБК направил запросы в правительства республик Марий Эл, Крым и правительства Пензенской, Владимирской и Брянской областей.
Пандемия COVID-19 потребовала перераспределения бюджетных ассигнований, для того чтобы устранить последствия распространения вируса. Это, в свою очередь, увеличивает риски обеспечения лекарствами орфанных пациентов в регионах, констатируют авторы доклада.
Федерализация — наиболее реальный механизм исправления ситуации, но альтернативой может стать механизм дофинансирования, считает директор центра социально-экономических исследований ЦСР Лора Накорякова. «Важно использовать бюджеты разного уровня, и в случае недостатка средств на полное обеспечение потребностей пациентов государством должны рассматриваться и другие источники, например средства из краудфандинговых сборов, которые сейчас очень слабо координируются», — сказала она РБК.
Причины проблем с диагностикой орфанных заболеваний
Кроме неравномерного распределения расходов, аналитики ЦСР выделяют и другие недостатки существующей системы.
В частности, когда на регион приходится относительно малое количество пациентов с конкретным заболеванием, он не может себе позволить закупку дорогостоящего диагностического оборудования и другой аппаратуры. Кроме того, значителен дефицит специалистов узкого профиля, которые могли бы выявить и контролировать ход лечения орфанных пациентов. А система диспансерно-динамического наблюдения, которая призвана отслеживать эффективность лечения наряду с эффективностью вложений в него, не используется почти в двух третях регионов.
Среди других причин поздней выявляемости заболеваний аналитики ЦСР называют недостаточную квалификацию врачей первичного звена, отсутствие у них мотивации к расширению знаний.
Решить вопрос с помощью специалистов федерального центра способна телемедицина, считает председатель экспертного совета Всероссийского общества орфанных заболеваний Екатерина Захарова. «Целесообразно более широкое использование телемедицины, а также финансирование транспортных расходов в качестве части процесса лечения», — говорит она.
Налицо и сложности с нормативно-правовой базой, отмечают аналитики ЦСР. База, которая позволяла бы эффективно вести регистр пациентов, по которому осуществляется лечение, сформирована не в полном объеме. Также, несмотря на приказ Минздрава № 1342н, согласно которому гражданин может выбрать медорганизацию за пределами своего региона, такая практика слабо распространена.
Опрошенные ЦСР специалисты в области орфанных заболеваний обращают внимание, что на федеральном уровне проблемы необеспеченности пациентов лечением или неготовности его финансировать не возникают. Тогда как на уровне регионов эти проблемы «относятся к числу наиболее острых». Эксперты отмечают также непрозрачность критериев включения и исключения препаратов в списки, по которым идет финансирование. В постановлении правительства № 871, которое регламентирует этот вопрос, указано, что одним из условий включения препарата в перечень является преимущество по сравнению с другими препаратами. «Однако не детализируются критерии определения этого преимущества», — говорится в докладе ЦСР.
В пресс-службе Минздрава в ответ на запрос РБК подтвердили, что в ряде регионов есть трудности с лекарственным обеспечением орфанных пациентов. В министерстве добавили, что по поручению президента «активно прорабатывают механизм» использования средств, которые будут получены после введения НДФЛ по ставке 15% для россиян, зарабатывающих свыше 5 млн руб. в год, в целях обеспечения оказания медпомощи и лекарственного обеспечения детям с тяжелыми заболеваниями, угрожающими жизни, и хроническими заболеваниями.
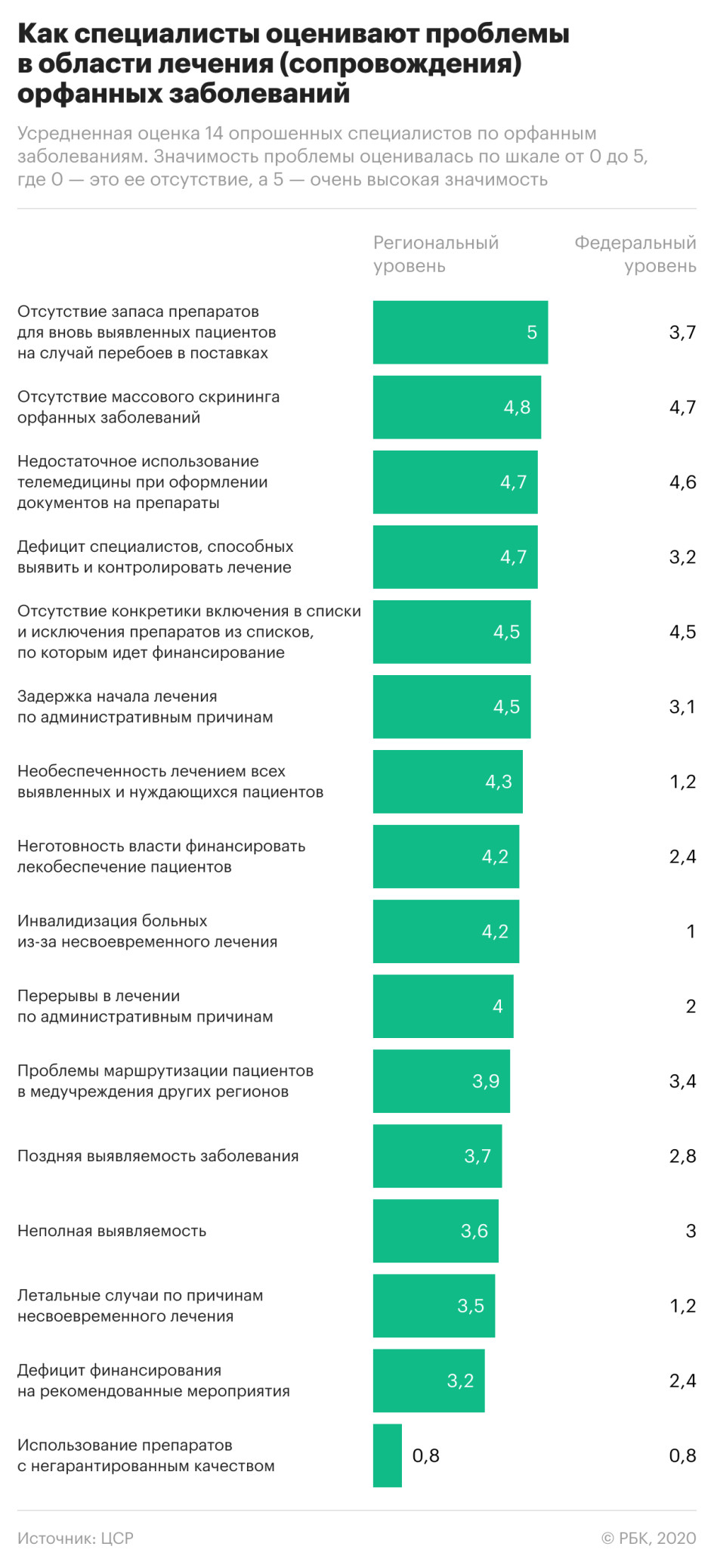
Поможет ли централизация закупок решению проблемы
После перевода пяти заболеваний на федеральный уровень в 2019 году средняя стоимость упаковки по шести препаратам, применяемым при терапии, снизилась на 12% в первый год, указывают на плюсы федерализации авторы исследования. Кроме того, охват орфанных пациентов лечением увеличился, и его проведение стало бесперебойным.
Эксперты по орфанным заболеваниям предлагают следующие механизмы федерализации расходов на закупку препаратов для страдающих орфанными заболеваниями:
- перенос всех расходов на федеральный бюджет;
- софинансирование расходов федеральным и региональными бюджетами;
- перенос на федеральный бюджет закупок только лекарств (за региональными сохраняются закупки лечебного питания).
«Важным обстоятельством в пользу внедрения механизма софинансирования региональных расходов из средств федерального бюджета является необходимость сохранения вовлеченности региональных властей в решение проблем орфанных пациентов. Как отмечалось выше, существует целый ряд проблем, которые не могут быть решены без активного участия местной власти», — утверждают эксперты.
При любом сценарии федерализации сообщество экспертов настаивает на том, что увеличение финансирования необходимо. С точки зрения специалистов, наиболее предпочтителен вариант поэтапной федерализации, когда на федеральный бюджет переводится часть из 17 угрожающих жизни заболеваний или, к примеру, наиболее проблемных регионов, поскольку в случае одновременного перевода всех заболеваний на федеральный уровень существует риск использования неподходящих препаратов. Кроме того, единовременная процедура перевода может стать стимулом для роста числа отечественных дженериков, прогнозируют эксперты.

По мнению аналитиков ЦСР, важнейшая задача для обеспечения орфанных пациентов — внедрение статистического учета, при котором появится возможность отслеживать эффективность назначаемой терапии. Федерализация позволит снизить многие риски, но для комплексного решения вопроса с орфанными заболеваниями необходимо выстраивание полноценной системы поддержки, которая бы включала меры по социализации пациентов, повышение квалификации врачей, актуализацию стандартов лечения и другие меры, заключают эксперты центра.
Ранее в Минфине РБК сообщали, что проект бюджета на будущие три года предусматривает выделение следующих средств на программу «14 ВЗН»: в 2020 году — 61,8 млрд руб., в 2021-м — 64,3 млрд руб., на 2022 год заложено 66,9 млрд руб. Также с 2021 по 2023 год будет ежегодно выделяться более 60 млрд руб. на лечение детей, страдающих редкими заболеваниями. Эти средства появятся после введения с 1 января 2021 года НДФЛ по ставке 15% для россиян, зарабатывающих свыше 5 млн руб. в год. При этом пациентские организации настаивают, что эти расходы должны быть существенно увеличены.
Источник


