- Путь лекарства
- Путь лекарства
- Конкурс «био/мол/текст»-2018
- Да здравствует идея!
- Операция «Мишень»
- На поиски лигандов
- Сокращай, оптимизируй!
- Тестировали, тестировали, да вытестировали
- Выходи на рынок!
- Новейшие методы разработки лекарственных средств
- Разработка и регистрация лекарственных средств
- Текущий выпуск
- ОТ РЕДАКЦИИ
- ПРАКТИЧЕСКИЕ РЕКОМЕНДАЦИИ
- МЕРОПРИЯТИЯ
- ПОИСК И РАЗРАБОТКА НОВЫХ ЛЕКАРСТВЕННЫХ СРЕДСТВ
- ФАРМАЦЕВТИЧЕСКАЯ ТЕХНОЛОГИЯ
- МЕТОДЫ АНАЛИЗА ЛЕКАРСТВЕННЫХ СРЕДСТВ
- ДОКЛИНИЧЕСКИЕ И КЛИНИЧЕСКИЕ ИССЛЕДОВАНИЯ
- РЕГУЛЯТОРНЫЕ ВОПРОСЫ
Путь лекарства
16 октября 2018
Путь лекарства
Создание нового лекарства требует большого количества ресурсов и времени. Невозможно предугадать успешность результата: подходящее, казалось бы, вещество, может дать сбой на любом этапе.
Автор
Редакторы
Инфографика на конкурс «био/мол/текст»: Казалось бы, для читателей «Биомолекулы» нет ничего понятнее, чем процесс создания лекарства. Однако почти никто не делал из этого инфографику — для смертных попроще. Вкратце — отсюда вы узнаете, сколько времени занимает процесс создания лекарства и насколько это недешево. И может быть, догадаетесь, что, если по телевизору сказали, что ученые обнаружили вещество, способное победить рак какую-нибудь заразу, то еще ох как рано бежать в аптеку в надежде купить новое лекарство.

Конкурс «био/мол/текст»-2018
 Эта работа заняла первое место в номинации «Наглядно о ненаглядном» конкурса «био/мол/текст»-2018.
Эта работа заняла первое место в номинации «Наглядно о ненаглядном» конкурса «био/мол/текст»-2018.
Генеральный спонсор конкурса — компания «Диаэм»: крупнейший поставщик оборудования, реагентов и расходных материалов для биологических исследований и производств.
Спонсором приза зрительских симпатий выступил медико-генетический центр Genotek.
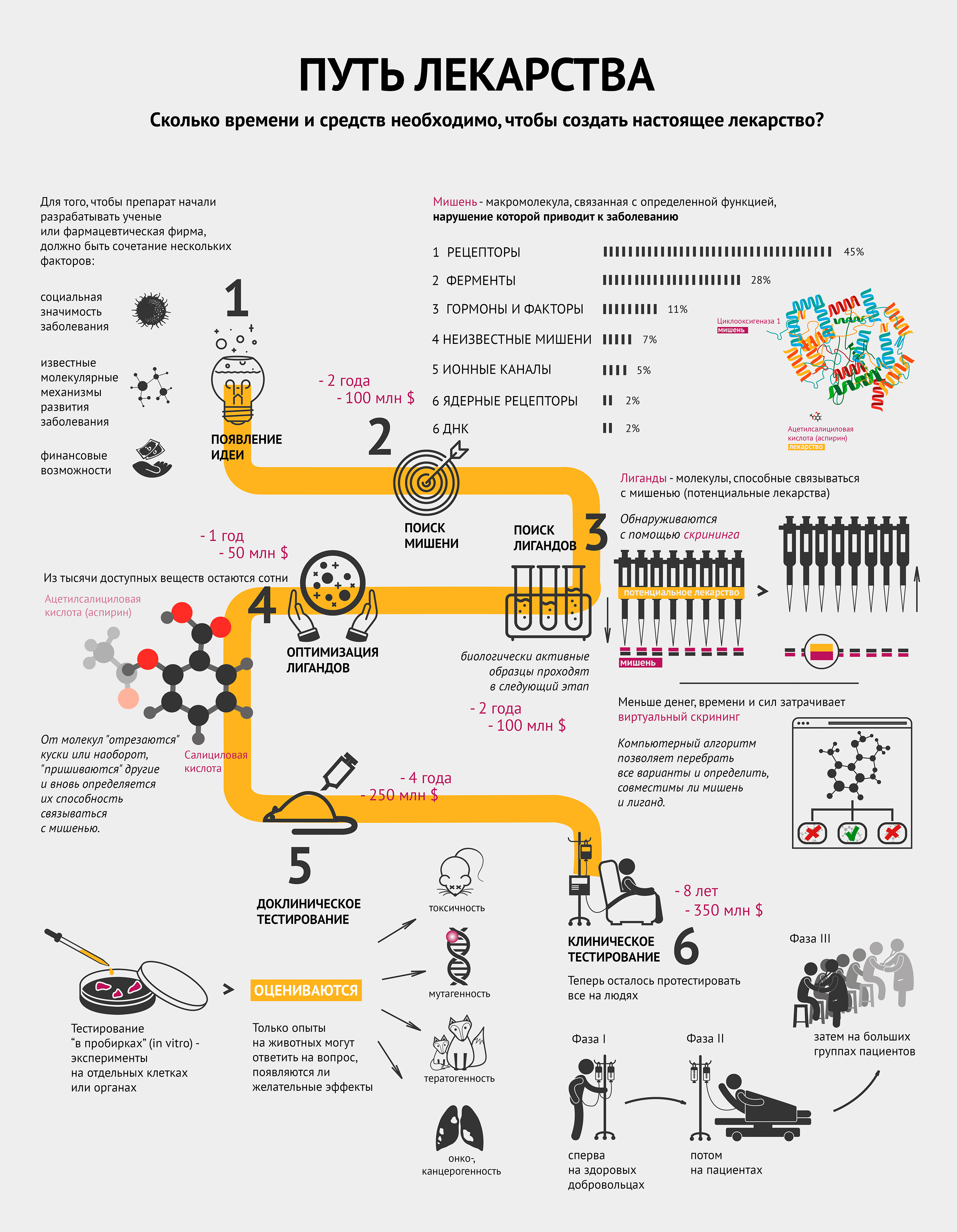
Да здравствует идея!
Для того чтобы препарат начали разрабатывать ученые или фармацевтическая фирма, должно быть сочетание нескольких факторов [1]:
- социальная значимость заболевания;
- известные молекулярные механизмы развития заболевания;
- финансовые средства и возможности по созданию конкретного лекарства.
Другими словами, должна появиться идея.
Операция «Мишень»
Совместными усилиями группа ученых выбирает мишень и способ воздействия на нее для лечения или предотвращения заболевания.
Мишень — это биологическая макромолекула, связанная с определенной функцией, нарушение которой приводит к заболеванию [2]. Чаще всего мишенями являются белки — рецепторы и ферменты. Инфографика демонстрирует, какие макромолекулы чаще всего становятся мишенями [2]. Забегая вперед, стоит отметить, что затем к мишени подбирают вещество — лекарство. Самый распространенный пример — циклооксигеназа 1 (мишень) и ацетилсалициловая кислота (аспирин) (лекарство) — тоже проиллюстрирован (см. также видео).
Видео. Лекция Валентина Табакмахера «Драг-дизайн. Современный подход к созданию лекарств».
На поиски лигандов
После того, как ученые нашли мишень, им нужно понять, чем в нее целиться. Лиганд (потенциальное лекарство) — это химическое соединение (как правило, низкомолекулярное), специфически взаимодействующее со своей мишенью и тем самым влияющее на процессы внутри клетки [2].
Исследование всех возможных веществ, конечно, нереально: существует не менее 10 40 лигандов. Поэтому на структуру потенциальных лигандов накладывают ряд ограничений, которые существенно сужают поиск. В качестве отправной точки обычно используют библиотеки соединений, которые создаются специализированными компаниями по условиям, заданным разработчиком, или уже имеются в арсенале фармацевтической фирмы. Такие библиотеки могут содержать миллионы веществ [3].
Воздействуют ли выбранные лиганды на мишень, помогает определить скрининг. Он бывает лабораторным (in vitro) или компьютерным (in silico). В случае с лабораторным скринингом на особые предметные стекла — плашки, содержащие в тысячах микролитровых лунок тестовую систему, например молекулы белка-мишени или целые клетки (при необходимости — генетически модифицированные), — робот раскапывает из пипеток исследуемые вещества, следуя заданной программе. Потом происходит считывание данных, говорящее о том, в какой лунке обнаружена биологическая активность. Детектор может определять ее по радиоактивному сигналу, флюоресценции, поляризации света и многим другим параметрам [3].
Сокращай, оптимизируй!
Из тысяч доступных веществ с более-менее определенными свойствами необходимо выбрать сотни молекул, способных после дальнейшей модификации и испытаний на бактериях или культурах клеток дать десятки так называемых кандидатных соединений, предназначенных для доклинических исследований, включая тестирование на животных.
Оптимизация может заключаться в «отсечении» части известного лиганда, или наоборот, добавлении к нему новых элементов и новой проверке на взаимодействие с мишенью. Возвращаясь к аспирину: он получился из салициловой кислоты путем добавления ацетильной группы.
Тестировали, тестировали, да вытестировали
Отобранные соединения сначала тестируются в биохимико-фармакологических исследованиях или экспериментах на клеточных культурах, изолированных клетках и изолированных органах. Так как эти модели не способны полностью воспроизвести весь комплекс биологических процессов в настоящем организме, любое потенциальное лекарство тестируется на животных. Только опыты на животных могут ответить на вопрос, появляются ли желательные эффекты в нетоксичных или малотоксичных дозах.
Исследование токсичности призвано оценить:
- токсичность при кратковременном и длительном применении;
- возможность генетических повреждений (генотоксичность, мутагенность);
- возможность развития опухолей (онко- и канцерогенность);
- возможность рождения больного плода (тератогенность).
На животных исследуемые соединения испытываются также на поглощение, распределение, метаболизм и выделение (фармакокинетика) [4].
После этого этапа отсева на стадию клинических испытаний на людях остается в лучшем случае 1−3 препарата (напомню, что изначально было примерно 1000 потенциальных лекарств!).
Выходи на рынок!
Клиническое тестирование включает в себя несколько фаз, которые иллюстрирует инфографика [5].
Сначала проводится исследование новых препаратов на здоровых лицах с целью определить, наблюдаются ли у человека эффекты, обнаруженные в тестах на животных, и выявить взаимоотношения между дозой и эффектом.
Потом потенциальный новый препарат апробируется на избранных пациентах для определения терапевтической эффективности при заболевании, для которого он предназначен. Положительное действие должно быть явным, а нежелательные эффекты приемлемо малы.
Далее к исследованию привлекаются большие группы пациентов, с помощью которых исследуемое лекарство сравнивается со стандартным лечением по исходам терапии [4].
В процессе клинических испытаний многие новые лекарства признаются негодными к применению.
Решение одобрить новый препарат принимает национальный регулирующий орган (в России — Фармкомитет МЗ РФ). Заявители (фармацевтические компании) представляют в регулирующий орган полный комплект документации преклинических и клинических испытаний, в которых полученные данные об эффективности и безопасности удовлетворяют установленным требованиям и предполагаемую форму выпуска продукта (таблетки, капсулы и т.д.)
После получения одобрения новое лекарство может продаваться под торговой маркой и, таким образом, становится доступным для назначения врачами и распространения в аптеках. Параллельно идет разработка технологического процесса производства лекарственного средства, требований к качеству, методов анализа.
По мере распространения препарата за ним продолжается наблюдение. Окончательное суждение о соотношении «польза—риск» нового лекарства может быть сделано только на основании долговременного опыта его применения. Таким образом, определяется терапевтическая ценность нового лекарственного препарата.
В разных случаях процесс разработки нового лекарства от идеи до реализации занимает примерно от 5 до 18 лет. Суммарная стоимость разработки, с учетом препаратов, не достигших рынка, часто превышает 1 млрд долларов (до 2,5 млрд в среднем) [6].
Источник
Новейшие методы разработки лекарственных средств
Разработка и регистрация лекарственных средств

Научно-производственный рецензируемый журнал «Разработка и регистрация лекарственных средств» — актуальное бесплатное прикладное издание и информационный портал для специалистов, задействованных в сфере обращения лекарственных средств. Журнал предназначен для фармацевтических предприятий-производителей и их сотрудников из отделов разработки, контроля качества, регистрации, производства и развития; сотрудников лабораторных центров, контрактно-исследовательских организаций, научных и образовательных учреждений. Включен в Перечень рецензируемых научных изданий, в которых должны быть опубликованы основные научные результаты диссертаций на соискание ученой степени кандидата наук, на соискание ученой степени доктора наук с 2015 года.
Включен в Перечень научных изданий, в которых должны быть опубликованы основные результаты исследований в рамках диссертаций по фармацевтическим наукам, представляемых к защите в диссертационных советах РУДН (от 01.10.2018 г.)
Основные пять тематических разделов журнала «Разработка и регистрация лекарственных средств» включают цикл развития лекарственного средства от его создания до получения регистрационного удостоверения. Первый раздел посвящен поиску и разработке новых лекарственных средств, второй — фармацевтической технологии и рассматривает научные и практические направления от разработки и производства исходных фармацевтических ингредиентов, технологий и оборудования – до создания стандартных и терапевтически эффективных лекарственных препаратов. Третий раздел описывает аналитические методики контроля качества; четвертый раздел посвящен подходам к оценке эффективности и безопасности лекарственных средств, проведению долклинических и клинических исследований; в пятом разделе рассматриваются вопросы валидации методик, подготовки регистрационного досье, жизненный цикл лекарственного препарата в GxP окружении.


Текущий выпуск
ОТ РЕДАКЦИИ
В этот раз ежегодная научно-практическая конференция состоялась в живом формате в Москве. Организаторами конференции выступили ООО «Центр Фармацевтической Аналитики» и научно-производственный журнал «Разработка и регистрация лекарственных средств». Мероприятие прошло 25 июня в Москве в Технопарк «КАЛИБР». Мероприятие было посвящено жизненному циклу биотехнологических лекарственных средств. Мы обсудили отдельные аспекты разработки, исследований и производства различных групп биотехнологических лекарственных средств, таких как моноклональные антитела, низкомолекулярные гепарины, инсулины, пептидные препараты и другие.
ПРАКТИЧЕСКИЕ РЕКОМЕНДАЦИИ
Точное и воспроизводимое количественное определение примесей в фармацевтических препаратах является важной частью разработки лекарств. Примеси должны быть доведены до уровня, который гарантирует, что качество продукта будет таким же хорошим или лучше, чем качество материалов, используемых в доклинических исследованиях безопасности и клинических испытаниях. Рекомендации Q3A и Q3B Международного Совета по Гармонизации (ICH) в отношении лекарственных веществ и фармацевтических препаратов обеспечивают нормативные ожидания в отношении исследования и контроля примесей, включая вещества, связанные с процессом, и продукты разложения. Пороговые значения для идентификации и оценки безопасности примесей могут основываться на относительных процентах или непосредственно в миллиграммах воздействия, в зависимости от природы лекарственных веществ и продуктов1. Таким образом, точная оценка уровней примесей необходима как для руководства разработкой, так и в течение всего жизненного цикла препарата.
Цель статьи: демонстрация прямого переноса аналитических методов ВЭЖХ из системы Agilent™ 1260 Infinity на платформу UltiMate 3000 и платформу Vanquish.
В статье показаны преимущества платформ UltiMate 3000 и Vanquish для прямого переноса ВЭЖХ методов, согласно требованиям USP.
- Гибкая регулировка общего объема системы в системах Thermo Scientific™ UltiMate™ 3000 и системах УВЭЖХ Thermo Scientific™ Vanquish™ упрощает перенос аналитических методов ВЭЖХ.
- Расширенные аппаратные функции системы ВЭЖХ Thermo Scientific™ Vanquish™ позволяют гибко регулировать общий объем задержки градиента системы для облегчения точной настройки во время переноса.
- Эквивалентные хроматографические результаты получены с обеими системами, но улучшенное разрешение и повторяемость системы обеспечиваются системой Thermo Scientific™ Vanquish™.
- Если чувствительность обнаружения является критической проблемой, то технология Thermo Scientific™ LightPipe™ является оптимальным решением.
МЕРОПРИЯТИЯ
Научная школа «Кадровый резерв для биофармацевтической и химической промышленности» традиционно проводится в Российском университете дружбы народов с целью ознакомления потенциальных абитуриентов с образовательными программами, реализуемыми в университете. Также задачей научной школы является представление результатов научной работы и обмен мнениями как между выдающимися учеными, так и молодыми учеными — студентами, магистрантами, аспирантами.
Первая международная фармацевтическая выставка-форум «ТехноФарм Сибирь» 2021 стала одним из масштабных событий очного формата в условиях пандемии коронавируса. Выставка-форум состоялась весной 2021 года и собрала более 30 компаний и 700 участников вместе. На мероприятии присутствовали региональные и федеральные игроки, эксперты из научного сообщества и фармацевтического производства, заместитель Министра промышленности, торговли и развития предпринимательства Новосибирской области Васильев Вадим Витальевич. Организатор выставки-форума — компания «ТехноФарм» и непосредственный директор — Екатерина Вастьянова.
С 5 по 8 октября в наукограде Кольцово Новосибирской области состоится международный научный конгресс в сфере биотехнологий, вирусологии и биофармацевтики OpenBio. Традиционно в его рамках пройдут научная конференция и бизнес-форум. Мероприятие организуют в гибридном формате: эксперты, спикеры и гости смогут участвовать онлайн.
ПОИСК И РАЗРАБОТКА НОВЫХ ЛЕКАРСТВЕННЫХ СРЕДСТВ
Введение. Заболевания печени при всем многообразии клинических проявлений имеют общие патогенетические звенья на клеточном уровне. Группа гепатопротекторных средств представлена лекарственными препаратами, проявляющими разносторонние механизмы защиты клеток печени от воздействия повреждающих факторов, основными из которых являются мембраностабилизирующее, антиоксидантное, регенеративное, детоксикационное, желчегонное и противовоспалительное действие. Высокая терапевтическая и гепатопротекторная эффективность современных лекарственных препаратов в большей степени обусловлена их метаболическими эффектами, а так же способностью связывать свободные радикалы и активные формы кислорода в клетке.
Текст. Цель настоящей работы заключается в формировании аналитического обзора литературных данных, посвященных вопросам ассортимента и концепциям совершенствования лекарственных форм гепатопротекторных средств. По данным анализа и систематизации современных публикаций, посвященных применению гепатопротекторов в терапии заболеваний печени, показана необходимость разработки новых составов и комбинаций биологически активных веществ с проявлением разносторонних механизмов гепатопротекции, а так же совершенствование состава и технологий изготовления имеющихся средств традиционной терапии. Одним из актуальных направлений в настоящее время является использование новых веществ в разработке традиционных и инновационных лекарственных форм. Продолжается поиск биологически активных молекул с антиоксидантной, антирадикальной и мембраностабилизирующей активностью, которые можно рассматривать в качестве эффективных гепатопротекторов. Неотъемлемой задачей фармацевтической разработки является создание биодоступных лекарственных средств, обладающих пролонгированным действием и минимальным проявлением побочных эффектов. Перспективным направлением в фармацевтической технологии является разработка инновационных препаратов направленного транспорта биологически активных молекул к пораженному органу.
Заключение. В результате анализа современных данных выявлены приоритетные направления разработки и совершенствования существующих составов, основанные на современных подходах к получению инновационных лекарственных форм. Показана актуальность совершенствования лекарственных форм гепатопротекторов, представленных на фармацевтическом рынке. Особый интерес представляет разработка инновационных систем адресной доставки с эффективными и безопасными гепатопротекторами в различных комбинациях, в том числе и на основе производных коричных кислот.
ФАРМАЦЕВТИЧЕСКАЯ ТЕХНОЛОГИЯ
Введение. В статье представлены результаты исследования физико-химических свойств сухого экстракта листьев оливы, стандартизованного по гидрокситирозолу, — биологически активному веществу, входящему в химический состав надземных частей оливкового дерева.
Цель. Разработать составы для изготовления таблеток с сухим экстрактом листьев оливы и провести стандартизацию методом ОФ-ВЭЖХ по содержанию гидрокситирозола.
Материалы и методы. Субстанция сухого экстракта листьев оливы, микрокристаллическая целлюлоза (Avicel® PH-112), Аэросил® (Aeroperl® 300 Pharma), Лудипресс® (Ludipress®), лактоза, крахмал картофельный, натриевая соль карбоксиметилированного крахмала, магния стеарат, прямое прессование, ВЭЖХ.
Результаты и обсуждение. Проведены исследования физико-химических свойств сухого экстракта листьев оливы. Выбраны вспомогательные вещества для опытных образцов. Разработаны составы для дальнейшего таблетирования. Проведен комплекс исследований полученных таблеток согласно ГФ XIV на соответствие показателей качества.
Заключение. Проведен обзор научных данных о биологических свойствах сухого экстракта листьев оливы, свидетельствующий о перспективе разработки лекарственных препаратов на его основе. Разработаны составы таблеток с сухим экстрактом листьев оливы и исследованы их показатели качества.
Введение. В связи с увеличением частоты выявления случаев туберкулеза, вызванных штаммами микобактерий, устойчивых не только к традиционным, но и недавно введенными в клинический оборот противотуберкулезным средствам актуальной является задача поиска и разработки новых лекарственных средств, способных эффективно подавлять мультирезистентные МЛУ и ШЛУ — штаммы M. tuberculosis. Одним из наиболее перспективных классов такого рода соединений являются трифтор-производные бензотиазинонов, и, в частности, соединение PBTZ169 (МНН макозинон). Однако макозинон обладает существенными особенностями физико-химических свойств, которые затрудняют разработку на его основе лекарственных форм для перорального применения. Он относится к классу IV по BCS и характеризуется низкой растворимостью, низкой липофильностью, выраженной зависимостью растворения от pH среды, очень низкой биодоступностью при приеме внутрь.
Цель. Обосновать целевой профиль, критические показатели качества и разработать прототип пероральной лекарственной формы с модифицированным высвобождением макозинона, позволяющей в максимальной степени реализовать его фармакологическую активность.
Материалы и методы. На основе фармацевтической субстанции макозинона гидрохлорида и различных вспомогательных веществ нарабатывали экспериментальные таблетки с дозировкой макозинона 500 мг. Оценивали влияние состава сред и добавляемых вспомогательных веществ на растворимость макозинона в различных биорелевантных средах, степень набухания в жидкости и степень мукоадгезии экспериментальных таблеток к слизистой желудка свиньи. Для оценки кинетики высвобождения активного вещества использовали метод ВЭЖХ.
Результаты и обсуждение. С учетом особенностей свойств макозинона обоснована целесообразность создания его гастроретентивных лекарственных форм с замедленным высвобождением активного вещества, механизм задержки которых в верхних отделах желудочнокишечного тракта обеспечивается за счет набухания таблеток и повышенной адгезии к слизистой желудка. Экспериментально испытаны различные образцы таблеток, в которых модификация высвобождения активного вещества и степень набухания и мукоадгезии варьировали за счет введения в состав формуляций различных вспомогательных веществ, в том числе известных набухающих и биоадгезивных матричных агентов.
Заключение. Наиболее перспективными для последующих фармакокинетических исследований признаны образцы высокодозированных (500 мг) набухающих и мукоадгезивных таблеток, созданных по технологии двухстадийной грануляции с включением в состав первичных гранул смеси макозинона и гидроксипропил-бета-циклодекстрина и последующим внесением в межгранульное пространство комбинаций растворимого и нерастворимого гидрофильных набухающих и мукоадгезивных матричных агентов (ГПМЦ, ГЭЦ, ПЭО).
МЕТОДЫ АНАЛИЗА ЛЕКАРСТВЕННЫХ СРЕДСТВ
Введение. Создание новых эффективных антибактериальных препаратов для лечения и профилактики гнойно-воспалительных заболеваний является актуальной задачей современной фармации. Активное применение в терапии гнойной инфекции находят химиотерапевтические средства из класса фторхинолонов, к которым относится офлоксацин.
Цель. Разработка способа количественного определения офлоксацина в комплексном препарате «Офлоксазоль».
Материалы и методы. Для проведения анализа использовали субстанцию офлоксацина, титансодержащий гель «Тизоль», растворы офлоксацина на 95%-м этаноле, кислоты хлористоводородной 0,01 моль/л, мазь под условным наименованием «Офлоксазоль», содержащую 0,5 % препарата в геле «Тизоль». Исследование проводили методом спектрофотометрии в ближней УФ-области.
Результаты и обсуждение. При изучении спектров поглощения установлено, что для количественного спектрофотометрического анализа офлоксацина рационально использовать область длин волн 275-320 нм (λmax = 294 нм). Статистическая обработка результатов анализа показала, что относительная погрешность количественного определения не превышает ±1,66 %. Чувствительность определения офлоксацина равна 0,245 мкг/мл при А(min) = 0,02. Разработанная методика валидирована. Подтверждена ее специфичность, линейность, правильность и прецизионность. По градуировочному графику определено содержание офлоксацина в мягкой лекарственной форме, оно находится в пределах 0,0483-0,0562 г, что соответствует допустимым отклонениям.
Заключение. Проведенные исследования позволили разработать и предложить способ количественного определения офлоксацина в мази «Офлоксазоль», полученной на титансодержащей основе. Способ позволяет проводить оценку качества изготовления лекарственной формы, в том числе устанавливать содержание препарата с ошибкой, не превышающей нормативные отклонения.
Введение. Лекарственные препараты производных хиназолина обладают широким спектром фармакологических свойств, что делает эту группу достаточно уникальной среди остальных классов гетероциклических соединений. Субстанция VMA-10-18, обладающая церебровазодилатирующим, антидепрессивным, анксиолитическим и ноотропным свойствами, может стать новым перспективным лекарственным препаратом. В связи с этим актуальной задачей является разработка методик стандартизации данной субстанции.
Цель. Разработка и валидация методики количественного определения родственных примесей в новой биологически активной субстанции VMA-10-18 (Хиназофен) методом ВЭЖХ.
Материалы и методы. Для разработки условий хроматографического анализа использовали высокоочищенную субстанцию 3-[2-(4-метоксифениламино)-2-оксоэтил]-хиназолин-4(3Н)-она, а также ее родственные примеси: примесь I [исходный незамещенный хиназолин-4(3Н)-он] и примесь II (4-метоксихлорацетанилид). В качестве растворителя использовали спирт этиловый 95 %. Хроматограф Dionex UltiMate 3000 (Dionex, США) со спектрофотометрическим детектором. Система сбора и обработки данных Chromeleon v.7. Подвижная фаза — смесь ацетонитрила и ортофосфорной кислоты. Анализ выполняли в изократическом режиме.
Результаты и обсуждение. Разработаны оптимальные условия хроматографирования субстанции VMA-10-18 и ее примесей. Установлено, что для четкого разделения пиков субстанции и примесей между собой подвижная фаза должна содержать ацетонитрил и ортофосфорную кислоту в соотношении 80 : 20. Валидацию разработанной методики проводили с учетом требования ГФ XIV издания и рекомендациям ICH. Подтверждена специфичность, линейность, правильность и прецизионность разработанной методики. Линейность и правильность методики определяли на 7 уровнях концентраций. Коэффициент корреляции превысил значение 0,99. Свободный член уравнения линейной зависимости (a) для обеих примесей был меньше его доверительного интервала (Δа), что доказывает отсутствие систематической погрешности методики. При определении показателя «Сходимость» рассчитанное относительное стандартное отклонение не превышало 2 %. При определении внутрилабораторной прецизионности рассчитывали t-критерий Стьюдента и F-критерий Фишера. Оба показателя отвечали заявленным требованиям.
Заключение. Разработана и валидирована методика количественного определения примесей в субстанции VMA-10-18 методом ВЭЖХ.
Введение. Липосомальные препараты обладают следующими преимуществами: защищают клетки организма от токсического действия лекарственных средств; пролонгируют действие введенного в организм лекарственного средства; защищают лекарственные вещества от деградации; способствуют проявлению нацеленной специфичности за счет селективного проникновения из крови в ткани; изменяют фармакокинетику лекарственных препаратов, повышая их фармакологическую эффективность; позволяют создать водорастворимую форму ряда лекарственных субстанций, повышая тем самым их биодоступность. Весьма актуальным является разработка липосомальных форм винпоцетина. В настоящее время при разработке состава липосомальных форм находит широкое применение методов молекулярного моделирования, которые являются удобным методом прогнозирования как свойств самих мембран, так и аспектов взаимодействия мембран с небольшими молекулами или белками.
Цель. Целью данного исследования является моделирование процесса сборки липосомы из фосфолипидов соевого лецитина в присутствии винпоцетина методом молекулярной динамики; а также прогнозирование распределения винпоцетина между внутренней полостью липосомы, фосфолипидной мембраной и дисперсионной средой по результатам моделирования.
Материалы и методы. Для моделирования процесса образования липосом был использован метод крупнозернистой молекулярной динамики в силовом поле Martini 2.2 с использованием программы Gromacs 2016.4. Сборка моделируемой системы — раствора фосфолипидов соевого лецитина в воде производилась с помощью интернет-сервиса Charmm-GUI->Inputgenerator->Martinimaker->Randombuilder.
Результаты и обсуждение. Результаты молекулярного моделирования показали, что молекулы винпоцетина не проникли внутрь липосомы, а адсорбировались на ее поверхности. Это связано с низкой растворимостью винпоцетина в гидрофобной среде мембраны липосомы соевого лецитина.
Заключение. Показано, что минимальный диаметр липосомы, образующейся из очищенного соевого лецитина, составляет 15,3 нм. Винпоцетин не проникает внутрь липосом из очищенного соевого лецитина, а адсорбируется на внешней поверхности их мембраны. Поверхностный избыток при этом по результатам моделирования крупнозернистой молекулярной динамики при температуре 298 К в спиртоводной среде составляет 1,2 • 10 -7 моль/м 2 .
Введение. Все больший научный интерес для практической онкологии вызывают производные индолокарбазола. В лаборатории химического синтеза Национального медицинского исследовательского центра онкологии им. Н.Н. Блохина синтезирован ряд N-гликозидов, индоло[2,3-а]карбазола под лабораторным шифром ЛХС. Одним из наиболее перспективных соединений этого класса является ЛХС-1208 — 6-амино-12-(α-L-арабинопиранозил)индоло[2,3-а]пирроло[3,4-c]карбазол-5,7-дион. По механизму биологического действия ЛХС-1208 относится к ингибиторам протеинкиназы С и представляет большой интерес для терапии злокачественных новообразований.
Цель. Химико-фармацевтическая стандартизация фармацевтической субстанции ЛХС-1208.
Материалы и методы. Лабораторные образцы фармацевтической субстанции ЛХС-1208. Методы исследования: гравиметрия, спектрофотометрия, поляриметрия, высокоэффективная жидкостная хроматография (ВЭЖХ), ядерная магнитно-резонансная (ЯМР) спектроскопия высокого разрешения и инфракрасная (ИК) спектроскопия.
Результаты и обсуждение. Оценку качества ЛХС-1208 проводили по показателям, принятым в XIV изданием Государственной фармакопеи Российской Федерации для контроля качества фармацевтических субстанций. ЛХС-1208 — оранжевый аморфный порошок, без запаха; растворим в диметилсульфоксиде (ДМСО) и диметилформамиде (ДМФА); очень мало растворим в спирте этиловом 95 % и практически нерастворим в воде. Подлинность субстанции подтверждается ЯМР- и ИК-спектрами, а также электронными спектрами поглощения. Значения удельного оптического вращения ЛХС-1208 (1 % раствор в ДМФА) укладываются в интервал от +58° до +61°. Все изученные образцы субстанции были свободны от неорганических примесей, сульфатной золы, тяжелых металлов и содержали не более 1,0 % воды, определенной методом титрования по К. Фишеру. Содержание возможных родственных примесей в субстанции ЛХС-1208 и содержание основного действующего вещества определяли методом ВЭЖХ. Исследованные лабораторные образцы фармацевтической субстанции ЛХС-1208 содержали не более 1,0 % любой единичной и не более 3 % суммы неидентифицированных примесей. Содержание основного действующего вещества составило более 97 %.
Заключение. В результате проведенной работы были отобраны критерии и параметры качества, а также разработаны методики их определения, позволяющие адекватно оценить качество и стандартность фармацевтической субстанции ЛХС-1208.
Введение. В настоящее время пристальное внимание в области фармации и медицины направлено на поиск новых источников биологически активных веществ различного происхождения, в том числе и растительного. В качестве перспективного источника рассматривается древесное растение Elaeagnus argentea. Несмотря на его широкое применение в пищевой промышленности, народной медицине в качестве противовоспалительного, общеукрепляющего, противомикробного средства, химический состав данного растения изучен недостаточно.
Цель. Качественно-количественное определение основных групп биологически активных веществ (БАВ) в сырье (листьях) Elaeagnus argentea, произрастающего на территории Астраханской области, для дальнейшей разработки методики стандартизации сырья данного растения.
Материалы и методы. Листья Elaeagnus argentea были заготовлены весной в восточной части дельты Астраханской области (Приволжский район). Сушка сырья производилась в естественных условиях. В исследовании были использованы унифицированные методики: для установления количества флавоноидов и сапонинов использовали спектрофотометрический метод, аскорбиновой кислоты -титриметрический метод. Для анализа флавоноидов в листьях Elaeagnus argentea в качестве экстрагента использовали водно-спиртовой раствор 70 % концентрации. Количественное содержание флавоноидов определяли в полученном экстракте сырья в пересчете на лютеолин-7-глюкозид. Количество сапонинов в листьях Elaeagnus argentea определяли в пересчете на олеаноловую кислоту. В качестве экстрагента использовали 96%-й этанол. Количественное определение аскорбиновой кислоты в водном извлечении измельченного сырья проводили методом титриметрии, основанным на способности восстанавливать 2,6-дихлорфенолиндофенол.
Результаты и обсуждение. При фармакогностическом изучении БАВ в листьях Elaeagnus argentea установлено содержание аскорбиновой кислоты не менее 0,32 %, суммы флавоноидов в пересчете на лютеолин-7-глюкозид — не менее 1,92 %; сапонинов — 2,38 %, что указывает на необходимость более детального исследования фитохимического состава других морфологических групп растения Elaeagnus argentea, произрастающего на территории Астраханской области.
Заключение. Полученные в ходе исследования данные могут быть использованы для подтверждения качества сырья (листьев) Elaeagnus argentea. Более детальное исследование листьев на наличие других групп БАВ позволит использовать полученные данные для разработки нормативной документации (НД) на лекарственное растительное сырье «Лоха листья».
ДОКЛИНИЧЕСКИЕ И КЛИНИЧЕСКИЕ ИССЛЕДОВАНИЯ
Введение. В ФГБНУ ВИЛАР разработан лекарственный препарат «Флакозид» обладающий гепатопротекторным действием. Препарат представлен в лекарственной форме — таблетки 0,1 г для приема внутрь.
Цель. Проанализировать клиническую эффективность и безопасность терапии флакозидом по клиническими лабораторным методам исследования, а также моторике желчного пузыря и желчевыводящих протоков у пациентов с заболеваниями гепатобилиарной системы.
Материалы и методы. Результаты клинических исследований флакозида (таблетки 0,1 г) проанализированы у 99 пациентов с хроническим активным гепатитом (ХАГ), хроническим бескаменным холециститом (ХБХ) и жировой дистрофией печени (ЖДП), проведенные в двух клинических учреждениях: ФГБОУ ВО «Пермский государственный медицинский университет им. академика Е. А. Вагнера» Минздрава России и ЦНИИ гастроэнтерологии. «Флакозид» назначали на фоне лечебной диеты (стол № 5) по 0,1-0,2 г 3 раза в день после еды в течение 32 дней и повторными курсами (3-5) на протяжении 6-12 месяцев. Анализ эффективности и безопасности флакозида проводили на основании результатов клинических и лабораторных исследований: общий и биохимический анализ крови, общий анализ мочи, электрокардиограммы (ЭКГ). Для изучения моторики желчного пузыря и желчевыводящих путей использовали методику многофракционного дуоденального зондирования (МФДЗ) с определением функционального состояния сфинктерного аппарата желчного пузыря и желчевыводящих путей. В пузырной и печеночной желчи определяли ее биохимический состав. Всем больным проводили рентгенологическое исследование желудочно-кишечного тракта, сканирование печени и гепатографию с йод-131-бенгал-роз.
Результаты и обсуждение. При ХАГ, ХБХ и ЖДП применение флакозида внутрь в суточных дозах 0,3-0,6 г в течение 25-45 дней приводило к улучшению общего состояния пациентов, уменьшению боли в правом подреберье, снижению диспептических расстройств, улучшению аппетита. По данным холецистографии улучшались показатели концентрационной и сократительной способности желчного пузыря. По степени выраженности лечебного эффекта «Флакозид» не уступал силибору и карсилу, а по некоторым показателям (улучшение моторики желчного пузыря и желчевыводящих путей) заметно их превосходил.
Заключение. Лечение флакозидом улучшало функциональное состояние печени, уменьшая синдром цитолиза и холестаза. «Флакозид» рекомендован в клинической практике в комплексном лечении заболеваний гепатобилиарной системы, таких как ХАГ, ХБХ и ЖДП.
Введение. Лаппаконитин представляет собой алкалоид, содержащийся в корнях и надземной части аконита белоустого (Aconitum leucostomum Vorosh.) и обладающий антиаритмическим действием. После приема препаратов лаппаконитина в организме человека образуется 8 изученных фармакологически активных метаболитов, наибольшую активность среди которых проявляет N-дезацетиллаппаконитин. Фармакокинетика препаратов лаппаконитина изучена недостаточно. Препараты лаппаконитина имеют узкий терапевтический диапазон и обладают большим количеством побочных эффектов, поэтому для оценки безопасности применения препаратов лаппаконитина возникает необходимость полноценного изучения его фармакокинетики.
Цель. Целью исследования является разработка методики определения лаппаконитина и его активного метаболита N-дезацетиллаппаконитина в плазме крови и в цельной крови человека методом высокоэффективной жидкостной хроматографии с тандемным масс-селективным детектированием (ВЭЖХ-МС/МС).
Материалы и методы. Определение лаппаконитина и N-дезацетиллаппаконитина в плазме крови и в цельной крови человека проводили методом ВЭЖХ-МС/МС. В качестве пробоподготовки был использован способ осаждения ацетонитрилом.
Результаты и обсуждение. Разработанная методика определения лаппаконитина и N-дезацетиллаппаконитина в плазме крови и в цельной крови человека была валидирована по следующим валидационным параметрам: селективность, эффект матрицы, калибровочная кривая, точность, прецизионность, степень извлечения, нижний предел количественного определения, перенос пробы, стабильность.
Заключение. Разработана и валидирована методика определения лаппаконитина и N-дезацетиллаппаконитина в плазме крови и в цельной крови человека методом ВЭЖХ-МС/МС. Подтвержденные аналитические диапазоны методики составили 0,50-50,00 нг/мл в биологической матрице для лаппаконитина и 0,50-100,00 нг/мл в биологической матрице для N-дезацетиллаппаконитина. Полученные аналитические диапазоны позволяют применять разработанную методику для проведения фармакокинетических исследований препаратов лаппаконитина.
РЕГУЛЯТОРНЫЕ ВОПРОСЫ
Введение. Настоящая публикация посвящена описанию последовательности проектирования и внедрения технологических процедур маркировки иммунобиологических лекарственных препаратов, выпускаемых ФКУЗ РосНИПЧИ «Микроб» Роспотребнадзора. В свете выполнения требований Федерального закона «Об обращении лекарственных средств» материалы данной статьи, несомненно, являются актуальным.
Текст. В статье представлена поэтапная последовательность внедрения в процесс производства лекарственных препаратов технологических процедур маркировки и взаимодействия с системой мониторинга движения лекарственных препаратов. На подготовительном этапе (этап № 1) решались следующие основные вопросы: проверка идентичности сведений о лекарственных препаратах в Государственном реестре лекарственных средств и в системе автоматической идентификации «ЮНИСКАН/ГС1 РУС»; определение способа и возможности нанесения средства идентификации на вторичную упаковку; внесение изменений в фармакопейные статьи предприятия на каждый вид препаратов. Этап № 2 (разработка требований к системе маркировки, сериализации, верификации и агрегирования) включал в себя следующие мероприятия: разработка функциональной модели процесса маркировки в ФКУЗ РосНИПЧИ «Микроб» и определение ответственных подразделений за реализацию данной схемы; определение способа упаковки во вторичную упаковку (ручной или автоматический), а также необходимой степени агрегации и требуемой автоматизации процесса, исходя из анализа функциональной модели и технологического процесса маркировки; анализ опыта внедрения систем маркировки лекарственных препаратов; анализ имеющейся IT-структуры ФКУЗ РосНИПЧИ «Микроб»; проведение мониторинга рынка производителей оборудования и программного обеспечения; разработка технических требований к создаваемой системе маркировки, сериализации, верификации и агрегирования. Этап № 3 (реализация на производственных участках системы маркировки, сериализации, верификации и агрегирования) включал в себя следующие мероприятия: поставка оборудования и проведение пуско-наладочных работ; квалификация оборудования (IQ/OQ); обучение персонала; внесение изменений в нормативные документы. В материалах, посвященных реализации заключительного этапа, рассмотрены вопросы валидации технологических процедур маркировки лекарственных препаратов и взаимодействия с системой маркировки, сериализации, верификации и агрегирования.
Заключение. Проведенные работы позволили производить лекарственные препараты в соответствии с требованиями Федерального закона «Об обращении лекарственных средств» и Постановления Правительства РФ от 14.12.2018 г. № 1556 «Об утверждении Положения о системе мониторинга движения лекарственных препаратов для медицинского применения». Изложенный материал может представлять интерес для производителей, выпускающих лекарственные препараты в небольшом объеме.
Введение. В технологии твердых лекарственных форм часто используют стадию влажного гранулирования. К наиболее сложному с инженерной и технологической точки зрения способу ее реализации можно отнести метод влажной грануляции в смесителе-грануляторе с высоким усилием сдвига. Смеситель-гранулятор имеет две мешалки, расположенные во взаимно перпендикулярных плоскостях. С помощью импеллера осуществляется смешивание сухих компонентов, а чоппер, включаясь при распылении увлажнителя, обеспечивает формирование гранулята. Задавая разную частоту вращения чоппера, можно получать гранулят разного размера. Данный гранулятор имеет следующие преимущества: обеспечивает высокий выход продукта (≥ 99 %), занимает небольшие рабочие пространства, а замкнутая конструкция обеспечивает защиту окружающей среды.
Цель. Цель исследования — масштабирование процесса гранулирования на примере технологии производства твердых лекарственных форм с использованием стадии влажного гранулирования в смесители-грануляторе с высоким усилием сдвига.
Материалы и методы. В качестве фармацевтической субстанции использовали сухой экстракт «N» и вспомогательные вещества: лактоза моногидрат, микрокристаллическая целлюлоза (МКЦ) (MICROCEL® MC-102), крахмал картофельный, повидон (Plasdone™ K-29/32), стеарат кальция. Гранулят в условиях масштабирования получали в смесителе-грануляторе с высоким усилием сдвига SMG3-6-1 (Chongqing Jinggong Pharmaceutical Machinery Co., Ltd., Китай). Технологические свойства сухого экстракта «N» и гранулята определяли по методикам, описанным в ГФ XIV.
Результаты и обсуждение. На основании проведенных экспериментальных исследований установлено, что полученный гранулят как и при получении в лабораторных условиях, так и в условиях масштабирования обладает хорошей сыпучестью, а также имеет однородный фракционный состав. Наличие чоппера в смесителе-грануляторе позволило получить более однородный фракционный состав гранулята. Увеличение загрузки в 10 раз не влияло на состав гранулята. При масштабировании процесса гранулирования был проведен анализ рисков, определены и структурированы факторы, влияющие на технологический процесс. Установлено, что наиболее важными являются стадии смешения и непосредственно гранулирование. Для получения однородной смеси была задействована дополнительная единица оборудования — смеситель.
Заключение. В результате масштабирования процесса гранулирования подобраны параметры проведения влажного гранулирования в смесителе-грануляторе (оптимальная загрузка, скорость вращения импеллера и чоппера), оценены технологические свойства полученного гранулята, риски, влияющие на технологический процесс, составлена причинно-следственная диаграмма (диаграмма Исикавы).
Источник


