- Надлежащие фармацевтические практики (Good Practice, GxP)
- Отраслевые стандарты серии GxP (GLP/GCP/GMP)
- GLP. Надлежащая лабораторная практика
- Правила GLP включают в себя:
- GСP. Надлежащая клиническая практика
- GMP. Надлежащая производственная практика
- GMP – надлежащая производственная практика
- «Американская комната страха»
- Сертификат GMP: подтверждение качества лекарственных средств
- К каким производствам применима эта процедура?
- Нормативная база
- Преимущества обладания сертификатом
- Стандарт GMP в международной практике
- Правила GMP в России
- Процедура получения сертификата в России
- Документы для сертификации
- Сроки сертификации
- Стоимость получения сертификата
Надлежащие фармацевтические практики (Good Practice, GxP)

Отраслевые стандарты серии GxP (GLP/GCP/GMP)
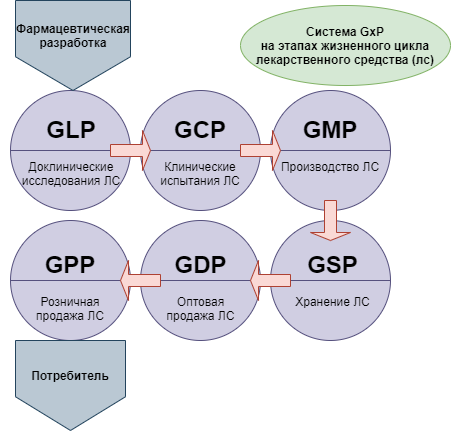
GxP (Good … Practice, Надлежащая … практика) — признанная во всем мире система обеспечения качества лекарственных средств. Система GxP охватывает все этапы жизненного цикла лекарственного средства, от фармацевтической разработки, испытаний, изготовления, хранения до использования конечным потребителем, а именно:
- Доклинические (лабораторные) исследования, которые регулируются правилами GLP (Good Laboratory Practice, Надлежащая лабораторная практика),
- Клинические испытания, которые регулируются правилами GCP (Good Clinical Practice, Надлежащая клиническая практика),
- Производство, которое регулируется правилами GMP (Good Manufacturing Practice, Надлежащая производственная практика),
- Хранение, которое регулируется правилами GSP (Good Service Practice, Надлежащая практика обслуживания, хранения),
- Оптовая торговля, которая регулируется правилами GDP (Good Distribution Practice, Надлежащая практика оптовой продажи),
- Розничная торговля, которая регулируется правилами GPP (Good Participatory Practice, Надлежащая практика розничной продажи).

Русский Регистр оказывает услуги по сертификации на соответствие стандартам серии GxP: практикам GLP, GCP и GMP.
GLP. Надлежащая лабораторная практика
GLP (Good Laboratory Practice, Надлежащая лабораторная практика) – система качества, охватывающая организационный процесс и условия, при которых выполняются неклинические исследования лекарственных средств, связанные со здоровьем и экологической безопасностью.
Правила GLP включают в себя:
- требования к организации испытаний;
- требования к личному составу исследователей;
- требования к помещениям, в которых проводятся испытания и содержатся животные;
- требования к качеству животных, к условиям их содержания и кормления;
- требования к лабораторному оборудованию и к его калибровке;
- требования к испытуемому и контрольному веществу;
- требования к составлению и проведению подробной стандартной методики экспериментальных работ и к порядку проведения испытаний;
- требования к регистрации данных и оформлению отчета;
- требования к службе контроля за качеством испытаний;
- стандартные методики экспериментальных работ.
Национальным аналогом GLP в РФ является стандарт ГОСТ 33044–2014 Межгосударственный стандарт «Принципы надлежащей лабораторной практики», текст которого идентичен GLP.
GСP. Надлежащая клиническая практика
GCP (Good Clinical Practice, Надлежащая клиническая практика) — международный этический и научный стандарт планирования и проведения исследований с участием человека в качестве субъекта, а также документального оформления и представления результатов таких исследований.
Правила GCP призваны обеспечить достоверность результатов клинических испытаний, а также безопасность и охрану прав и здоровья людей, принимающих участие в данных испытаниях в качестве субъектов.
Национальным аналогом GCP в РФ является стандарт ГОСТ Р 52379–2005 «Надлежащая клиническая практика», текст которого идентичен GСP.
GMP. Надлежащая производственная практика
GMP (Good Manufacturing Practice, Надлежащая производственная практика) – международный стандарт, который устанавливает требования к производству и контролю качества лекарственных средств для человека и животных, а также специальные требования к производству активных фармацевтических субстанций и отдельных видов лекарственных средств. Стандарт GMP регулирует и оценивает параметры производства и лабораторной проверки.
Национальным аналогом GMP в РФ является стандарт ГОСТ Р 52249–2009 «Правила производства и контроля качества лекарственных средств», текст которого идентичен GMP.
Источник
GMP – надлежащая производственная практика
Международный стандарт GMP (Good Manufacturing Practice, Надлежащая производственная практика) – система норм, свод правил и указаний, способствующих обеспечению качественного производственного процесса, в том числе хранения и испытаний продукции.

Этот стандарт включает множество показателей, которым должны соответствовать предприятия, выпускающие ту или иную продукцию. Его главное отличие от процедуры контроля качества, где исследуются выборочные образцы продуктов и обеспечивается пригодность лишь этих самих образцов (возможно, их партий), GMP отражает целостный подход и регулирует, оценивает собственно параметры производства и лабораторной проверки.
GMP нашел широкое применение в фармацевтической промышленности, при производстве микроэлектронных устройств, в высокотехнологичных отраслях промышленного изготовления продуктов питания, упаковочной, оптической продукции, сенсорных устройств, медицинской техники и пр.
Так, в комплексе со стандартами GLP (Good Laboratory Practice) – надлежащая лабораторная практика и GCP (Good Clinical Practice) – надлежащая клиническая практика GMP стандартизирует некоторые аспекты качества медицинского обслуживания населения.
Помимо этого в мировой практике распространены и другие стандарты надлежащих практик исследования, производства, дистрибуции, хранения и лекарственного обеспечения: GEP (Good Engeneering Practice) – надлежащая инженерная практика; GSP (Good storage practices for pharmaceuticals) – надлежащая практика хранения фармацевтической продукции; GPP (Good Pharmacy Practice) – надлежащая аптечная практика; GDP (Good Distribution Practice) – надлежащая дистрибуторская практика.
«Американская комната страха»
Начало настоящей борьбы за качество пищевых продуктов и лекарственных средств (ЛС) положили США в самом начале прошлого века – в 1906 году их конгресс принял Закон о доброкачественности пищевых продуктов и медицинских препаратов. Этот закон поспособствовал созданию одного из первых государственных контрольных органов в этой области, известного сейчас как FDA (Управление по санитарному надзору за качеством пищевых продуктов и медикаментов) и также разрешал конфискацию нелегальных (фальсифицированных) пищевых продуктов и ЛС.
В 1933 году FDA организовало выставку, названную «Американской комнатой страха» и представляющую продукты, косметику, медицинские устройства и лекарственные препараты, смертельно опасные для человека. Среди экспонатов было устройство для поддержания матки (контрацептив) и прокалывающее ее при неправильном введении; препарат для похудания, неизбежно приводящий к смерти пациента; средство для удаления волос, вызывающее облысение; косметические средства на основе ртути, свинца; оральный «Эликсир сульфаниламида» на диэтиленгликоле, погубивший к тому времени 107 человек. Выставка, потрясшая многих американцев, привела к принятию нового более жесткого Закона о пищевых продуктах, медикаментах и косметике (1938), требующего от производителя подтверждения безопасности их продукции до ее появления в продаже и расширившего полномочия FDA (до остановки производства и уголовного преследования).
Далее – трагедия 1941 года. Около 300 человек, погибших и пострадавших от приема сульфатиазоловых таблеток (в их состав входил фенобарбитал), подтолкнули FDA к пересмотру требований к производству и контролю над качеством ЛС, а конгресс – к новому Закону «О здравоохранении», значительно приблизивших мир к системе GMP.
А следующий шаг к системе стандартов качества был связан с еще более драматическими событиями. Имя им – талидомид. Этот препарат на фармрынке Европы был представлен как средство от бессонницы и для избавления от утренней тошноты беременных. При его регистрации регулирующие учреждения и не предполагали насколько мощным тератогенным эффектом обладает это лекарство. Число врожденных уродств у детей, рожденных матерями, принимавших препарат, насчитывало 10 тысяч. С этого времени новые строгие поправки в законодательство сделали обязательным подтверждение эффективности и безопасности ЛС в клинических исследованиях (до апробирования на людях – испытания на животных).
Без сомнения, человечество по-настоящему выстрадало свой путь к правилам GMP. Конечно же, фармацевтические скандалы продолжали будоражить общественное мнение. Были смерти от приема тайленола (отравленные капсулы ацетаминофена) и отзыв с рынка 31 млн упаковок этого популярного безрецептурного препарата. Был скандал с дженериковыми препаратами и недобросовестными сотрудниками FDA, получавших взятки. Но каждый из них вел к совершенствованию законодательства и стандартов GMP (1965, 1971, 1978, 1987, 1992 гг.).
И проблемы качества ЛС касались далеко не только США. Фармацевтический рынок во второй половине XX века развивался стремительно, приобретая глобальный характер и все сильнее ощущая потребность в формировании международных стандартов, позволяющих унифицировать, регламентировать производство, хранение и распространение ЛС. Активными инициаторами появления и продвижения правил GMP стали ответственные производители, не желавшие мириться с многочисленными конкурентами, не желавшие вкладывать значительные средства в систему обеспечения качества лекарств; новые компании, стремящиеся к цивилизованному рынку и эффективным вложениям капитала; страховые компании, умеющие профессионально считать деньги.
И вот история GMP, начавшаяся в США в 1963 году появлением на свет первых правил безопасного и качественного изготовления ЛС (стандартная форма официального документа принята в 1968 г.), получила поддержку международных экспертов. В 1968 году появился первый международный документ по GMP, разработанный специалистами Всемирной организации здравоохранения. А через год была принята резолюция ВОЗ, рекомендующая применять правила GMP всем странам (1969). Восемь государств именно так и поступили. Сегодня национальные правила GMP имеются более чем в 40 странах. Помимо этого есть региональные правила GMP, правила GMP стран Евросоюза, стран-участников «соглашения по фармакологическому контролю», стран– членов ассоциации, стран юго-восточной Азии, международные правила GMP. По данным ВОЗ, в настоящее время свыше 140 стран участвуют в системе удостоверения качества медикаментов в международной торговле, основанной на соблюдении правил GMP.
Но тогда, в 1969 году, наша страна не поддержала инициативу ВОЗ. Минздрав СССР заявил о своей незаинтересованности во внедрении GMP – готовился собственный документ, предполагавший стандартизацию оборота ЛС по международным правилам. В 1974 году он появился (рекомендательные правила производства лекарств РТМ 64-7-81-74, пересмотренные в 1981 г. – правила ОМУ 64-33-81), надолго отложив вопрос перехода к GMP.
В 1991 году Европейский союз Директивой 356/91 утвердил новые правила GMP (GMP EU), для стран, входящих в него (в 2003 г. внесены изменения). В том же году Советский Союз попытался гармонизировать свое законодательство с международной практикой. Но только после его распада страны СНГ самостоятельно приступили к созданию нормативной базы по примеру международных.
Российский стандарт GMP начал готовиться с 1998 года (по аналогии с GMP EU). Правительством РФ была утверждена федеральная целевая программа «Развитие медицинской промышленности на 1998-2000 гг. и на период до 2005 г.», предусматривающая поэтапное внедрение требований GMP на российских фармацевтических предприятиях до 2006 г. В 2004 году появился ГОСТ
52.249-2004 «Правила производства и контроля качества ЛС».
Отечественные принципы GMP для фармацевтической отрасли регламентированы национальным стандартом ГОСТ Р 52249-2009 «Правила производства и контроля качества лекарственных средств» (утвержден и введен в действие приказом Федерального агентства по техническому регулированию и метрологии от 20 мая 2009 г. № 159-ст). Стандарт распространяется на все категории ЛС и прописывает общие требования к их изготовлению и контролю качества, а также конкретные требования к производству активных фармацевтических субстанций и отдельных видов лекарственных препаратов. Надо заметить, что GMP – это не добровольный, а обязательный набор правил, поэтому он подлежит проверке государством. В нашей стране инспектирование производителей ЛС на соответствие стандарту GMP проводит Федеральное бюджетное учреждение «Государственный институт лекарственных средств и надлежащих практик».
В соответствии с Федеральным законом № 61-ФЗ «Об обращении лекарственных средств» в 2014 году все национальные компании, занимающиеся производством ЛС, обязаны были перейти на стандарт GMP, но сегодня только несколько десятков предприятий смогли привести свое производство в соответствие с требованиями государственного стандарта качества.
Постановлением Правительства РФ от 3 декабря 2015 года №1314 утверждены «Правила организации и проведения инспектирования производителей лекарственных средств на соответствие требованиям правил надлежащей производственной практики, а также выдачи заключений о соответствии производителя лекарственных средств указанным требованиям ».
В мае 2017 года национальные рынки обращения лекарственных средств пяти государств Евразийского экономического союза (ЕАЭС — Россия, Беларусь, Казахстан, Кыргызстан, Армения) объединились и начали работать в формате единого пространства и на основе общих правил надлежащей производственной практики Евразийского экономического союза.
Источник
Сертификат GMP: подтверждение качества лекарственных средств

Сертификат GMP — это соблюдение изготовителем лекарственных препаратов требований надлежащей производственной практики. В России они сформулированы в национальном стандарте ГОСТ Р 52249-2009, который идентичен правилам, действующим в Европейском Союзе.
К каким производствам применима эта процедура?
В настоящее время в странах, которые контролируют соответствие стандарту GMP на своих территориях, его правила применяются для проверки качества следующих категорий продукции:
- лекарственные препараты;
- медицинские изделия различного назначения, включая те из них, которые применяются в диагностических целях;
- продукты питания и ингредиенты для их производства;
- биологически активные добавки.
При этом для понимания ситуации следует принимать во внимание, что новая версия сертификации GMP — это не единственная система требований, которые в международной практике применяются в целях стандартизации медицинского обслуживания населения. Кроме них, производителям, работающим в такой сфере как фармация, необходимо соответствовать требованиям комплекса правил, объединенных под общим наименованием GxP:
- GLP — Good Laboratory Practice (надлежащая лабораторная практика);
- GCP — Good Clinical Practice (надлежащая клиническая практика);
- GDP — Good Distributon Practice (надлежащая дистрибьюторская практика);
- GACP — Good Agricultural and Collection Practice (надлежащая практика культивирования и сбора лекарственных растений).
Нормативная база
В Российской Федерации получение сертификата GMP осуществляется на основании действующей нормативной базы, включающей следующие основные правовые акты:
- национальный стандарт РФ ГОСТ Р 52249-2009, устанавливающий правила изготовления и контроля качества лекарственных препаратов;
- постановление Правительства от 5 июня 2008 года N 438 с рядом изменений, внесенных за последние годы, которое утверждает полномочия Министерства промышленности и торговли в этой области;
- постановление Правительства от 3 декабря 2015 года N 1314, устанавливающее порядок оценки соответствия производителей требованиям стандарта надлежащей практики;
- приказ Минпромторга от 14 июня 2013 года N 916, утверждающий правила применения надлежащей производственной практики в соответствии с актуальным стандартом;
- приказ Минпромторга от 26 мая 2016 года N 1714, определяющий административный регламент предоставления государственной услуги по выдаче документации, подтверждающей соответствие изготовителя установленным нормам надлежащей производственной практики;
- приказ Минпромторга России от 17.12.2015 N 4119, утверждающий правила ведения реестра сведений о том, какие лекарства имеют сертификат качества GMP в России.
При этом необходимо принимать во внимание, что в настоящий момент наша страна вместе с другими государствами, входящими в состав Евразийского экономического союза, находится на этапе становления общего рынка, объединяющего фармацевтическое и косметическое производство в границах Союза. Это предполагает в том числе введение в действие единых требований к качеству и безопасности таких продуктов. В соответствии с принятым в мире порядком они реализуются в форме внедрения стандартов надлежащей производственной практики. Применение таких стандартов регулируется следующими нормативными документами:
- Решение Совета ЕЭК от 3 ноября 2016 года N 77, утверждающее правила надлежащей производственной практики в границах ЕАЭС;
- Приказ Минпромторга от 4 сентября 2020 года N 2945, которым введен административный регламент предоставления госуслуги по выдаче документации, подтверждающей соответствие производств установленным правилам.
Обратите внимание!
Для полноценного применения разработанного административного регламента необходимо решение Правительства о порядке реализации некоторых процедур, связанных с проведением фармацевтических инспекций. Приказ № 2945 вступит в силу только после принятия соответствующего постановления: пока этого не произошло.
Преимущества обладания сертификатом
Несмотря на необходимость проведения достаточно сложной и дорогостоящей процедуры, производители знают, что сертификация по стандартам GMP является весьма важной для представителей фармацевтической отрасли. В частности, оно обеспечивает продукции и производству следующие серьезные преимущества:
- стабильное качество продукции, не зависящее от внешних факторов;
- повышение доверия потребителей, включая крупных оптовых покупателей, которые всегда отслеживают, какие производители имеют сертификат соответствия GMP на их продукцию;
- возможность вывода продукции на международные рынки, где ее может купить гораздо больше потребителей;
- возможность привлечения инвесторов для реализации проектов по расширению производства;
- получение преимуществ при участии в конкурсном отборе поставщиков, в том числе для государственных закупок.
КОММЕНТАРИЙ ЭКСПЕРТА АТТЭК
Каков срок действия сертификата?
Срок действия российских сертификатов составляет 3 года. При этом срок действия иностранного сертификата GMP составляет от 1 до 3 лет. По истечении этого периода сертификацию потребуется проходить заново. Кроме того, это означает, что на протяжении всего этого срока компании необходимо обеспечить соответствие своего производства и продукции требованиям комплекса правил GMP.
Кто в России занимается сертификацией по стандартам GMP?
Сейчас сертификация контролируется департаментом развития медицинской и фармацевтической промышленности Министерства промышленности и торговли РФ. Он является организацией, ответственной за обеспечение надлежащего контроля за качеством, безопасностью и эффективностью лекарственных средств. Осуществлением требуемых сертификационных процедур занимается Государственный институт лекарственных средств и надлежащих практик (ФГБУ «ГИЛС и НП»).
Стандарт GMP в международной практике
Процесс сертификации на соответствие лекарственного препарата стандартам GMP в международной практике имеет комплексный характер, а ее основной целью является подтверждение безопасности и действенности продукции. В этой связи для достижения поставленной цели специалисты аккредитованных сертификационных организаций не ограничиваются оценкой ряда выборочных образцов лекарственных препаратов, как это часто предусматривается другими стандартами. В процедуру установления требуемого уровня качества лекарств любой международный центр сертификации лекарственных средств включает оценку предприятия, занимающегося его выпуском. В результате эксперты, занимающиеся проведением сертификации, анализируют конкретный препарат и процесс его выпуска в следующих областях:
- оценка производства на соответствие критериям безопасности, включая проведение его проверки в отношении вероятности попадания в продукт посторонних примесей и веществ;
- оценка производства на соблюдение технических требований к выпуску продукции, включая выполнение условий относительно влажности, температуры и других параметров в производственных помещениях;
- оценка качества, безопасности и действенности лекарственных средств, производимых на конкретном предприятии;
- оценка соответствия параметров производства и характеристик лекарственного средства нормативной документации, принятой в рамках процедуры GMP.
Правила GMP в России
Порядок и сроки проведения всех операций в рамках этой процедуры, список лиц и организаций, ответственных за их осуществление, размер платы за проведение экспертной оценки и другие аспекты выполнения сертификации определены постановлением Правительства № 1314.
Процедура получения сертификата в России
Первым шагом для производителя, который желает пройти сертификацию, является подача соответствующего заявления в Минпромторг. В течение 10 рабочих дней специалисты ведомства проводят проверку корректности представленных в заявлении сведений и определяют возможность проведения сертификации.
В случае необходимости они вправе запросить у заявителя дополнительные документы, которые он обязан предоставить в течение 20 рабочих дней. В случае, если в отношении данного препарата принято положительное решение о проведении процедуры сертификации, необходимые данные направляются в ФГБУ «ГИЛС и НП», который в течение 20 рабочих дней с момента их получения обязан определить дату проведения сертификационных мероприятий и внести ее в график. Такая дата должна наступить не позднее 160 рабочих дней со дня, когда специалисты Минпромторга приняли положительное решение о сертификации, а сама экспертиза и расшифровка ее результатов должны занимать не более 10 рабочих дней.
На подготовку итогового отчета по результатам ее проведения исполнителю отводится 30 рабочих дней, а на его направление заявителю — 3 рабочих дня. Копия такого отчета также направляется в Минпромторг. На основании отчета формируется окончательное заключение, которое в случае положительного характера сопровождается выдачей сертификата производителю лекарственного препарата.
Документы для сертификации
Чтобы получить сертификат GMP в России, производитель обращается в уполномоченный орган с заявлением, к которому прилагает пакет документов, включающий:
- копию документа, подтверждающего наличие у заявителя полномочий по взаимодействию с контролирующей организацией;
- копия основного досье используемого производственного объекта;
- информация о фактах несоответствия препарата действующим требованиям к качеству и безопасности и о фактах отзыва медикамента из оборота за период не менее 2 лет;
- полный список лекарств, который изготавливаются на данном производственном объекте;
- копия лицензии на производство лекарств;
- письмо о согласии на проведение инспекции производства.
Важнейшие документы предоставляются заявителем в копиях, поскольку при утере их восстановить невозможно или очень сложно. Правила регламентируют, что если заявление подает иностранный производитель, и некоторые документы в составе пакета представлены на другом языке, они должны быть переведены на русский язык и заверены в установленном порядке.
Сроки сертификации
Общая продолжительность процедуры сертификации складывается из следующих сроков.
Этап сертификационной процедуры
Максимальная допустимая продолжительность
Проверка полноты пакета документации, представленной с заявлением о сертификации, и правильности ее оформления, назначение инспекции
10 рабочих дней
Направление информации о назначении инспектирования в уполномоченное учреждение, которое проводит проверку
Инспектирование и анализ лекарственного средства
160 рабочих дней
Принятие решения о выдаче заключения по результатам инспекционного отчета
10 рабочих дней
160-дневный период инспектирования включает внесение производителя в график инспекций, ожидание процедуры и проведение самой инспекции. Она должна занимать не более 10 рабочих дней.
Такой порядок действует, если в документации, поданной производителем, не обнаружат ошибок и недочетов, из-за которых ее могут направить на доработку. В этом случае вся процедура займет немногим более 180 рабочих дней, то есть свыше 8 месяцев.
Стоимость получения сертификата
Обязательной для всех производителей лекарственных средств, претендующих на получение сертификата, подтверждающего соответствие их продукции стандартам GMP, является оплата государственной пошлины за рассмотрение соответствующего заявления в Министерстве промышленности и торговли. Ее размер составляет 7500 рублей. Оплатить данную сумму необходимо еще до подачи заявления в ведомство, а ее размер никак не зависит от результатов рассмотрения документа.
Однако данная пошлина — это далеко не единственный и не самый крупный платеж, который потребуется осуществить производителю лекарств. Другой значительной статьей расходов станет плата за проведение экспертной оценки производства и продукции заявителя. Такая процедура выполняется специалистами ФГБУ «ГИЛС и НП»: для каждого из них предварительно проводится аттестация эксперта по GMP в России.
При этом размер платы за проведение оценки не является строго установленным, а определяется в зависимости от объема, характера и сложности необходимых процедур в соответствии с положениями приказа Министерства промышленности и торговли Российской Федерации от 11.01.2016 № 9 «Об утверждении методики определения размера платы за оказание услуги по инспектированию GMP». В случае, если проверка потребует проведения значительного объема работы и привлечения большого количества высококвалифицированных экспертов, размер платы за ее проведение может превышать 2,5 миллиона рублей.
Источник


