- Как тестируют лекарства?
- Обзор
- Подберите мне лекарство
- Опыты на животных
- Испытания на добровольцах
- Другие статьи по темам:
- Последняя проверка
- Как талидомидовая трагедия изменила мир
- Дизайны клинических исследований: как разрабатываются и какими бывают
- Этапы клинических исследований
- Этика клинических испытаний: информированное согласие
- Кто и как контролирует проведение клинических испытаний
- Будущее клинических испытаний
Как тестируют лекарства?
Обзор
Морфин, героин, эфедрин, мышьяк и свинец — не так давно эти вещества продавались как лекарства и назначались в качестве средств от кашля, боли или воспаления. Тестировать их сразу in vivo пришлось детям, беременным женщинам и остальным пациентам. О побочных эффектах тогда задумывались мало, альтернативных лекарств не было.
Сейчас от появления нового вещества, перспективного в плане решения каких-либо медицинских проблем до его регистрации в качестве лекарственного препарата проходит около 10-12 лет. За это время вещество подвергается сотням тестов и экспериментов. Но и после обретения торгового названия и своего места на аптечной полке лекарство многократно проверяют.
Современное тестирование лекарств обеспечивает защиту от токсичных, фальсифицированных и неэффективных препаратов. Но главная цель исследований лекарственных препаратов — поиск новых полезных свойств, расширение показаний, открытие новых лекарственных субстанций для лечения тех болезней, которые пока остаются не побежденными.
Подберите мне лекарство
Откуда берутся новые лекарства? Нередко их получают простым подбором «на заказ». Мишенью действия большинства фармпрепаратов являются белковые рецепторы или ионные каналы в оболочках клеток. Меняя структуру мишени, лекарство вызывает ряд желаемых эффектов. Если мы знаем строение рецептора, подобрать к нему «ключ» — дело техники. С помощью виртуального моделирования и роботов-лаборантов современная техника решает этот вопрос быстрее человека. Как это происходит?
После того, как определена точка приложения будущего лекарства, компания-разработчик приобретает подходящую под свои требования библиотеку химических веществ, которая может содержать сотни и тысячи различных соединений, обладающих похожими свойствами. Вещества тестируют, добавляя в тысячи микропробирок, где содержатся одинаковые белки-мишени или культуры клеток. Пробирки, где замечена биологическая активность, отбирают для дальнейших экспериментов и постепенно выделяют десяток наиболее перспективных прототипов для будущего лекарства. Их отправляются на стадию доклинических испытаний. Перебор вручную занял бы несколько человеческих жизней. С помощью работающей без перерывов и выходных роботизированной пипетки и компьютерного анализа поиск новых лекарств значительно ускоряется.
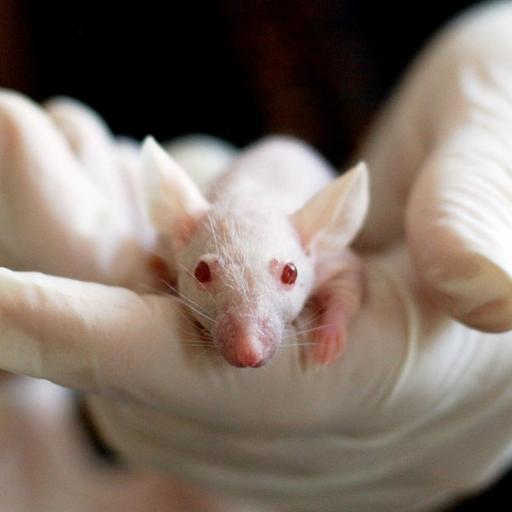
Опыты на животных
Доклинические испытания — это масштабный этап исследования кандидатов в лекарства. Он может длиться до 10 лет и проводится преимущественно на животных. Некоторые тесты, например при исследовании мутагенности, традиционно выполняют на культурах клеток, бактериях, мухах дрозофилах или аквариумных рыбках. В продвинутых лабораториях отдельные эксперименты научились моделировать в виртуальной реальности. Однако основные испытуемые — это грызуны: мыши, крысы, хомяки, морские свинки и кролики. Ряд тестов требует участия более крупных млекопитающих: собак, обезьян, реже — мини-пиги, кошек, хорьков и др.
Исследование проводится на здоровых животных обоих полов, отобранных по возрасту, размерам и происхождению. Испытуемых содержат в стандартизованных условиях, кормят и поят строго по режиму одинаковой пищей. Если будущее лекарство предназначено для детей, то исследования проводят на детенышах, лекарства для беременных испытывают на беременных самках, препараты геронтологической практики — на стареющих животных.
Цель доклинических исследований:
- Отсеять вещества, непригодные для дальнейших испытаний из-за токсичности, канцерогенности или других свойств.
- Определить «стартовую» безопасную дозу препарата для использования в первой фазе клинических исследований на людях.
- Оценить соотношение польза/риск для каждого кандидата в лекарства.
Важнейший аспект доклинических испытаний — проверка вещества на вредность. Начинают с определения дозы: нормальной переносимой, токсической и летальной. Определяют, какие органы и ткани животных оказываются более подвержены действию препарата? Какие побочные эффекты вызывает вещество?
Сначала препарат вводят с короткими 3-6-часовыми промежутками в течение суток и отслеживают результаты острой токсичности. Затем приступают к тестированию хронической токсичности. Эта часть исследования зависит от предполагаемой продолжительности лечебного курса у человека и может длиться от нескольких недель до года и более.
Далее испытывают влияние препарата на фертильность у самцов и самок животных. Определяют эмбриотоксичность и фетотоксичность — отрицательное влияние вещества на эмбрион, плод, а также отслеживают рост и развитие детенышей после рождения. Отдельно изучают мутагенность — способность вызывать мутации у взрослого животного и канцерогенность — вероятность злокачественных опухолей.
Сведения о канцерогенности вещества обязательно необходимы для получения разрешения на клинические исследования в нашей стране. Лекарства, которые будут продаваться без рецепта или предназначенные для длительного использования тестируются с особой тщательностью. Например, гормональные препараты проходят тестирование на канцерогенность около 7-10 лет, и подопытными, помимо крыс и мышей, должны быть собаки или обезьяны.
Данные проверки на вредность являются опорными для последующих экспериментов. Существуют коэффициенты пересчета доз исходя из массы тела. Ориентируясь на эти цифры можно проецировать влияние препарата на человека.
Второй аспект доклинических исследований — фармакокинетика — путь, который проделывает вещество в организме: как всасывается, как распределяется, вступает ли в химические превращения, какими органами и с какой скоростью выводится. Знание фармакокинетики дает представление о достижимой концентрации вещества в крови, а значит, позволяет точнее подобрать эффективную дозу для человека, выбрать оптимальную форму введения препарата в организм: через рот или с помощью инъекций.
Третий вопрос, на который отвечают лабораторные изыскания — предположительная эффективность кандидата в лекарства. Методология этой части исследования значительно отличается в зависимости от целевой сферы применения вещества. Например, при тестировании нейролептиков у животных исследуют их поведенческие реакции и двигательную активность. Для тестирования сердечных лекарств проводят испытания на собаках, кошках или обезьянах, у которых искусственно нарушают кровообращение сердца, имитируя стенокардию.
На территории России принят единый стандарт проведения доклинических исследований — ГОСТ 33044-2014, который соответствует международному стандарту GLP (Good Laboratory Practice — «Надлежащая лабораторная практика»). Только успешно прошедшие все фазы доклинических исследований кандидаты допускаются к изучению на людях, в среднем это 2 вещества из 100, остальные отбраковываются как слишком вредные или неэффективные. С учетом этих цифр трудно переоценить значение лабораторных животных в медицинской науке.
Испытания на добровольцах
«По-взрослому» будущие лекарства испытывают на здоровых и больных людях, которые должны добровольно выразить свое согласие на участие в эксперименте после того, как ознакомятся со всеми «за» и «против». Для здоровых людей аргументом «за» чаще является финансовая заинтересованность — за участие в клинических исследованиях можно заработать в среднем 15-20 тысяч рублей.
Здоровых добровольцев привлекают на клинические исследования 1 фазы. Группа подопытных здесь не более 100 человек. Людям дают различные дозы препарата и динамически определяют его концентрацию в крови, отслеживают побочные действия и их связь с дозой. Таким образом уточняют максимальную безопасную дозировку у человека. На этом этапе отсеиваются около 30% веществ.
Быстрым и дешевым видом клинического испытания является исследование на биоэквивалентность. Такие тесты применяют для регистрации дженериков — копий оригинальных лекарственных препаратов. Исследования биоэквивалентности проводят на небольшой группе из 12-24 здоровых добровольцев. Испытуемым вводят новый препарат и в течение 1-2 дней берут пробы крови с определенным интервалом. После измерения концентрации вещества в крови строят график и сравнивают его с таковым у оригинального лекарства. Успешные испытания биоэквивалентности позволяют считать схожими данные о токсичности и эффективности воспроизведенной копии и оригинального препарата. Таким образом, производитель дженерика не тратит время и деньги на полный цикл исследования лекарственного препарата. Все, что нужно — это доказать идентичность оригинальному лекарству.
2 фаза клинических испытаний — исследование на группе больных из 100-200 человек. Люди с различными заболеваниями обычно становятся участниками испытаний по рекомендации врача. Медицинское обследование на протяжении эксперимента, все виды лечения по профилю исследования в этом случае бесплатные. Для некоторых пациентов участие в клинических испытаниях — это шанс отстоять свою жизнь у смертельного заболевания, так как получить еще незарегистрированный и потенциально более эффективный лекарственный препарат другим путем невозможно.
Во время 2 фазы препарат используют в терапевтических дозировках и оценивают его эффективность, а также выявляют оставшиеся незамеченными ранее побочные эффекты. Требования к качеству 2 фазы значительно выше. В идеале это рандомизированные двойные слепые исследования с плацебо-контролем. Рандомизированные — значит всех участников делят на группы случайным образом, а не по желанию врача. Одна из групп будет получать вместо лекарства пустышку — плацебо. Двойное слепое — значит ни пациенты, ни врачи, участвующие в эксперименте, не будут знать о том, кто какое лечение получает. Для этого плацебо и традиционные препараты должны выглядеть одинаково, а данные о них и пациентах шифруются. Если препарат оказался эффективнее плацебо, лекарство допускают до следующего этапа. Отсев на этом этапе — около 70% вступивших в него веществ.
3 фаза — самая ответственная для кандидата. Успешно прошедшие его получают регистрацию в качестве лекарственного препарата. В 3 фазе участвуют от нескольких сотен до 3000 людей с соответствующим заболеванием. Главная цель — сравнить эффективность вещества с уже имеющимися препаратами или методами лечения. Исследование, как и на предыдущей фазе, должно соответствовать рандомизированному с двойным ослеплением. Заключительный этап испытаний преодолевают в среднем не более 30% кандидатов. Однако и после регистрации лекарства продолжают тестировать.
Каждый участник исследования подписывает договор, в котором указаны все риски, а также условия эксперимента. Организация, которая проводит клинические исследования обязана застраховать жизнь и здоровье каждого участника исследования за собственный счет. Сумма страховки составляет от 300 тысяч до 2 млн в случае смерти.
4 фаза — это исследования зарегистрированного препарата, который уже используется в качестве лекарства. Положительные результаты 4 фазы клинических испытаний обычно используются фирмой производителем в маркетинговых целях, отрицательные часто умалчиваются и не публикуются. Пострегистрационные исследования не являются обязательными. Они необходимы, когда есть потребность расширить показания для применения лекарства, если обнаружились его дополнительные полезные свойства. Иногда компании-производители проводят дополнительные исследования в пострегистрационном периоде для того, чтобы привести досье препарата к единому международному стандарту. То есть выполняют те исследования, которые ранее были необязательны или невозможны.
В нашей стране проведение клинических исследований стандартизировано по ГОСТ Р 52379-2005, что соответствует международному стандарту GCP (Good Clinical Practice — «Надлежащая клиническая практика»).
Фармкомпании далеко не всегда занимаются процедурой тестирования лекарств самостоятельно. Нередко они выступают лишь в роли заказчика, а исполнителем являются сторонние исследовательские организации, которые обеспечивают весь цикл от лабораторных испытаний до регистрации лекарства или только отдельные этапы этого процесса, что ускоряет рождение нового лекарства в среднем на 30%. Являются ли результаты исследований, выполненных с помощью аутсорфинга, более объективными — вопрос. Ведь исполнитель находится в прямой финансовой зависимости от заказчика.
После регистрации лекарства постоянно подвергаются контролю со стороны Росздравнадзора. Для проверки выборочно изымаются образцы препаратов прямо на производстве (для отечественных средств), при пересечении границы (для импортных лекарств), а также непосредственно в аптеках и больницах. Эксперты контролируют условия производства и хранения лекарств, проверяют подлинность документации, соответствие упаковки установленным правилам, а также исследуют состав лекарственных препаратов обычными химическими методами и с помощью спектроскопии.
Препараты, которые официально признаны лекарствами на территории нашей страны, проходят серьезную проверку перед выходом на фармацевтический рынок и после. Чем солиднее возраст лекарственного средства, тем лучше изучено его влияние на здоровье человека, тем выше его безопасность. Однако по эффективности старые лекарства нередко уступают вновь зарегистрированным, чьи отдаленные «побочки» нам еще предстоит испытать на себе.
Другие статьи по темам:
Все материалы сайта были проверены врачами. Однако, даже самая достоверная статья не позволяет учесть все особенности заболевания у конкретного человека. Поэтому информация, размещенная на нашем сайте, не может заменить визита к врачу, а лишь дополняет его. Статьи подготовлены для ознакомительных целей и носят рекомендательный характер. При появлении симптомов, пожалуйста, обратитесь к врачу.
Источник
Последняя проверка
Как устроены клинические исследования новых лекарств?
Несмотря на все достижения современной науки, невозможно предсказать, как новый лекарственный препарат подействует на организм человека, опираясь только на данные экспериментов на животных. Опыты на биологических моделях дают приблизительное представление о безопасности изучаемого лекарства. Клинические исследования (КИ) на людях-добровольцах — единственный инструмент получения доказательств эффективности и безопасности новых лекарств.
Сегодня кажется, что проверять лекарства на людях, соблюдая строгие правила, естественно и разумно. Но так было не всегда. Совместно с биотехнологической компанией BIOCAD рассказываем, когда КИ стали краеугольным камнем фармакологии, как их проводят и кто контролирует — и попытаемся представить, какое будущее ожидает клинические испытания в XXI веке.
Как талидомидовая трагедия изменила мир
Талидомид «открывали» дважды. В 1952 году действующее вещество (фталимидоглутаримид) разработали в швейцарской фармкомпании Ciba (сейчас она входит в концерн Novartis), но сочли бесперспективным. Два года спустя сотрудники немецкой компании Grünenthal случайно синтезировали его снова — как побочный продукт при поиске противовирусного препарата. На сей раз вещество все-таки запатентовали и начали исследовать его свойства.
Будущее лекарство протестировали по множеству показаний: от местного обезболивающего до антиконвульсанта. В итоге выяснилось, что лучше всего талидомид работает как успокоительное средство. Оставалось только подтвердить безопасность препарата, чтобы затем наконец выпустить его на рынок.
В середине XX века клинические испытания на людях уже проводились. Первое полноценное клиническое испытание по протоколу, очень похожему на современный, провели в 1954 году — тогда испытывали вакцину против полиомиелита, больше известную как вакцина Солка. Однако в 1950-1960-х «проверка на людях» еще не была обязательным этапом, который должен был преодолеть новый препарат, прежде чем попасть на рынок.
Разработчики талидомида решили, что для подтверждения безопасности вполне достаточно испытаний на самых популярных лабораторных животных: по одним данным это были мыши, по другим — крысы. Этого оказалось достаточно, чтобы в 1956 году немецкие власти одобрили новое успокоительное лекарство, в том числе для беременных женщин. Сомнений ни у кого не возникло: в то время считалось, что лекарственные препараты из материнской крови не могут попасть к плоду.
К 1960 году талидомид продавался уже в 46 странах — причем примерно в таких же объемах, как современный аспирин. Итог известен: 40 тысяч пострадавших от полинейропатии (поражения периферических нервов) и примерно 10 тысяч детей, которые родились с фокомелией (тяжелыми дефектами конечностей). Из этих детей выжило чуть больше половины. Все они остались инвалидами.
Талидомидная катастрофа полностью изменила методы тестирования лекарств. После этой истории выход новых препаратов на рынок перешел под контроль государства. Были разработаны правила проведения клинических исследований, единые для всех фармацевтических компаний. Важную роль в создании этих правил сыграла сотрудница Управления по санитарному надзору за качеством пищевых продуктов и медикаментов (FDA), доктор Фрэнсис Келси. Невзирая на давление фармкомпании и работодателя, с 1957 по 1962 год она не пускала талидомид в США. Именно ее «анти-талидомидные» статьи стали основой требований к современным клиническим испытаниям лекарств.
Талидомидовый скандал внес коррективы и в доклинические испытания препаратов. Исследования, проведенные уже после отзыва лекарства с рынка, показали, что животные разных видов могут по-разному реагировать на одно и то же действующее вещество. По до сих пор не до конца понятной причине мыши оказались менее чувствительными к талидомиду, чем приматы и кролики. Именно поэтому современные доклинические испытания должны включать проверку действующего вещества и на клеточных культурах «в пробирке», и на животных нескольких видов.
Дизайны клинических исследований: как разрабатываются и какими бывают
Согласно определению Всемирной организации здравоохранения (ВОЗ), клиническое испытание (КИ) — это любая научно-исследовательская работа, в ходе которой проспективно (то есть в соответствиями с критериями, изложенными в протоколе исследования — прим. ред.) набранные люди или группы людей подвергаются одному или нескольким вмешательствам, связанным со здоровьем, для оценки воздействия этих вмешательств на состояние здоровья.
По типу дизайна большую часть клинических исследований можно разделить на две большие группы: обсервационные (наблюдательные) и экспериментальные.
Во время обсервационных исследований исследователи активно не вмешиваются и только собирают данные, наблюдая за естественным развитием процесса. Такие исследования часто используют для анализа — они помогают понять, есть ли причинно-следственная связь между воздействием (например, лекарством) и результатом (например, выздоровлением).
Для изучения новых препаратов в подавляющем большинстве случаев используют экспериментальные исследования. Однако не все клинические исследования направлены только на изучение свойств препарата. Так, например, поисковые исследования (exploratory studies) позволяют получить общие данные по какому-либо предмету и служат базой для формирования гипотезы — которая, в свою очередь, будет проверена в ходе дальнейших более специфических испытаний. Поисковые исследования проводятся не чтобы доказать, что лекарство работает — а если требуется информация, критически важная для планирования дальнейшей клинической разработки препарата и проведения исследования в целом.
В этой статье мы будем описывать экспериментальные клинические исследования — так можно назвать любой эксперимент, в котором специально отобранных участников разделяют на две группы: экспериментальную, которая получает лекарство, и контрольную, которая получает либо плацебо — похожий по внешнему виду препарат, но без действующего вещества, либо другой лекарственный препарат с известными параметрами безопасности и эффективности.
Клинические исследования позволяют оценить эффект лекарства на здоровье. При этом все экспериментальные исследования отличаются друг от друга в зависимости от поставленных целей и задач. Это значит, что у них нет единого протокола клинического испытания — для каждого эксперимента план и дизайн проведения приходится создавать заново.
О чем фармкомпании говорят с учеными
Консультации с научным сообществом — важный этап планирования клинических исследований. Такие обсуждения проводятся до начала исследования еще на этапе подготовки документов. Без специалистов сотрудники фармацевтических компаний не смогут учесть важные нюансы из конкретной терапевтической области.
Специалисты могут подсказать, какие именно факторы необходимо учесть при стратификации, как часто нужно оценивать тот или иной показатель для формирования адекватных выводов, что именно важно в терапии для пациентов. Сотрудники фармакологических компаний обязательно спрашивают у экспертов, какие плюсы или минусы те отмечают для уже применяющихся лекарственных средств.
Протокол исследования — документ, который помогает экспериментатору планировать и анализировать этапы клинического испытания. Фактически это заранее сформированный и утвержденный план исследования, который помогает планировать время и бюджет на каждом этапе. Согласно Федеральному закону от 12.04.2010 № 61-ФЗ «Об обращении лекарственных средств», без правильно составленного протокола исследования нельзя получить разрешение на проведение клинического испытания. А поскольку уже утвержденный протокол без серьезных оснований менять запрещено, нужно очень тщательно его продумать.
В протоколе исследования прописывают:
- цели — что именно исследователи стремятся доказать в этом конкретном эксперименте. Как правило, цель звучит так: «Доказать эффективность и безопасность препарата»;
- место проведения эксперимента — на базе каких клиник в какой именно стране будет проводиться КИ;
- размер выборки пациентов — сколько будет участников в экспериментальной и контрольной группе;
- критерии включения и исключения пациентов — какими особенностями должны обладать пациенты, чтобы их включили в исследование;
- этические вопросы — важно, чтобы участники осознавали, что они принимают участие в эксперименте, осознавали возможные последствия и делали это добровольно;
- какие процедуры планируется использовать и как часто их нужно будет применять;
- методы обработки и анализа полученных данных — это позволяет убедиться, что результаты были зафиксированы и обработаны верно, и на их основании был сделан корректный вывод.
В протоколе прописаны важнейшие характеристики эксперимента: будет ли исследование слепым или открытым, рандомизированным или нерандомизированным, контролируемым или неконтролируемым. Каждая из этих характеристик помогает минимизировать «человеческий фактор» — то есть уменьшить предвзятость со стороны экспериментаторов и участников исследования.
Слепой метод подразумевает, что ни врачам, ни пациентам-участникам исследования неизвестно, какое именно лечение получали участники эксперимента. «Заслеплять» можно участников исследования, врачей и ученых, которые будут анализировать результаты.
Рандомизация — случайное и бессистемное распределение пациентов на группы. Рандомизация гарантирует, что каждый участник с одинаковой вероятностью может получить исследуемый препарат или контроль. «Заслепление» и рандомизация защищают эксперимент от искажений, связанных с личными предпочтениями врача-исследователя.
Вместе с рандомизацией может выполняться стратификация — уравновешивание контрольной и экспериментальной групп. Стратификация позволяет определить совместный эффект двух (и более) экспериментальных воздействий — это нужно, если, например, в процессе экспериментального лечения предполагается давать пациентам таблетки и ставить капельницы. Такой подход позволяет понять, что работало лучше — только таблетки, только капельницы или и то и другое одновременно.
Контролируемое клиническое испытание — исследование, при котором группу, получившую изучаемый препарат, сравнивают с контрольной группой. Как правило, люди, попавшие в группу контроля, получают уже применяемый в клинике лекарственный препарат с изученными свойствами — ведь речь идет о реальных пациентах, оставлять которых без лечения неэтично. Это может быть препарат аналогичного класса, разрешенный для применения у целевой популяции. А если его не существует — стандарт терапии, или (иногда) плацебо.
Какой именно контроль подойдет для конкретного исследования, зависит от особенностей пациентов с различными заболеваниями. Это определяется рекомендациями, которые создают регуляторные органы в сотрудничестве с ведущими врачами-экспертами по интересующему заболеванию. Рекомендации содержат указания, какой именно дизайн и контроль требуется использовать, а также какие именно параметры необходимо оценивать при определенной патологии.
На сегодняшний день стандартом испытаний для финального доказательства безопасности и эффективности лекарственного средства является рандомизированное, двойное слепое, контролируемое клиническое исследование. Только такой эксперимент позволяет достоверно установить, что между приемом лекарства и выздоровлением существует причинно-следственная связь.
Итак, чтобы зарегистрировать препарат и вывести его на рынок, требуется определенный план доказательства эффективности и безопасности препарата. Реализация этого плана требует времени. Полный цикл клинической разработки препарата, включая клинические испытания, в среднем занимает более 10 лет. Тем не менее, из каждого правила бывают исключения: в некоторых обстоятельствах возможна ускоренная регистрация препарата.
В 1992 году FDA разработала требования для ускоренной регистрации лекарственныхсредств. Согласно этим требованиям, ускоренная регистрация возможна для препаратов, предназначенных для терапии тяжелых заболеваний с дефицитом вариантов терапии. Чаще всего «ускоряют» регистрацию онкологических препаратов. Один из недавних и широко известных примеров — препарат пембролизумаб для лечения меланомы, который ускоренно одобрили по семи показаниям. При этом в России похожий препарат — пролголимаб — был зарегистрирован по стандартной процедуре..
Однако процесс ускоренной регистрации не отменяет необходимости исследования параметров, которые в норме должны изучаться на поздних фазах клинических исследований. В случае процедуры регистрации на условиях, которая предусмотрена Правилами регистрации и экспертизы лекарственных средств для медицинского применения, утвержденными решением Совета Евразийской экономической комиссии от 3 ноября 2016 г. N 78, регулятором может быть в качестве одного из дополнительных требований предусмотрено проведение пострегистрационных исследований. Если их результаты оказываются неудовлетворительными, регистрацию могут отменить.
В нашей стране Министерство здравоохранения может разрешить зарегистрировать препарат по ускоренной процедуре, принятой в рамках Постановления Правительства РФ от 3 апреля 2020 года № 441, в случае если он предназначен для применения в условиях угрозы возникновения, возникновения и ликвидации чрезвычайной ситуации. В этом постановлении говорится, что «сокращение объема . экспертиз» допустимо «в условиях угрозы возникновения и ликвидации чрезвычайной ситуации». Именно так зарегистрировали российский препарат Авифавир (МНН — фавипиравир), предназначенный для борьбы с коронавирусной инфекцией, и препарат Илсира® (МНН — левилимаб), который применяют для лечения осложненного течения коронавирусной инфекции.
Левилимаб — человеческое (не искусственное) моноклональное антитело против рецептора интерлейкина-6 (ИЛ-6). ИЛ-6 — это провоспалительный цитокин, то есть вещество, играющее ключевую роль в «запуске» некоторых аутоиммунных заболеваний. Именно поэтому препарат первоначально предназначался для лечения ревматоидного артрита. Клинические исследования первой и второй фазы доказали его безопасность и эффективность.
Однако интерлейкин-6 не только играет важную роль в патогенезе ревматоидного артрита, но и является ключевым агентом угрожающего жизни состояния — «цитокинового шторма». Так называется осложнение новой коронавирусной инфекции — тяжелая гипервоспалительная реакция, которую вирус провоцирует в организме некоторых людей.
Препарат для лечения осложнений коронавируса необходим срочно, поэтому сотрудники компании BIOCAD решили пересмотреть клиническую разработку препарата Илсира® (МНН: левилимаб) и чтобы препарат как можно быстрее попал в клиники и пациентам, решили инициировать дополнительное клиническое исследование 3 фазы, в которое включили пациентов с тяжелым течением COVID-19. Исследование на настоящий момент продолжается. В мае 2020 препарат Илсира® (МНН: левилимаб) зарегистрировали по новому показанию: в качестве средства для экстренной помощи при угрожающем жизни остром респираторном дистресс-синдроме, сопровождающемся повышенным высвобождением цитокинов.
Пример от BIOCAD
Дизайн клинического испытания: препарат Эфлейра®
Что за лекарство: BCD-085 (торговое наименование — Эфлейра®), или моноклональное антитело к интерлейкину 17. МНН — нетакимаб.
Зачем нужно лекарство: Препарат предназначен для лечения псориаза, псориатического артрита и анкилозирующего спондилита, который в нашей стране часто называют болезнью Бехтерева. Болезнь Бехтерева — воспалительный артрит, поражающий позвоночник и крупные суставы, при котором на месте воспаления образуются костные выросты, постепенно вызывая сращение костей — анкилоз.
Интерлейкин-17 — это провоспалительный цитокин, то есть «провокатор» воспаления, которое сопровождает аутоиммунные заболевания, в том числе спондилит. Исследователи предположили, что моноклональные антитела к этому веществу будут «отключать» воспаление — а, значит, и лечить аутоиммунные (иммуно-воспалительные) заболевания.
Код клинического исследования: BCD-085-5/ASTERA
Важнейшие характеристики клинического исследования: Международное многоцентровое рандомизированное двойное слепое плацебо-контролируемое клиническое исследование эффективности и безопасности препарата BCD-085 (ЗАО «БИОКАД», Россия). Этот тип дизайна лучше всего отвечает рекомендациям и правилам проведения клинических исследований.
Цель исследования: Доказать эффективность и безопасность препарата BCD-085 (нетакимаб) для лечения пациентов с анкилозирующим спондилитом.
Место и время проведения исследования: Российская Федерация: 19 государственных клиник; Республика Беларусь: 2 государственные клиники. Если в исследовании участвует несколько клиник, легче набрать нужное число участников, которые четко соответствуют критериям включения пациентов.
Размер выборки: 228 участников: 114 пациентов в опытной группе и столько же — в контрольной группе. Достаточный размер выборки рассчитывался специально для данного исследования.
Критерии включения пациентов: В исследование включались пациенты мужского и женского пола в возрасте 18-65 лет, с подтвержденным диагнозом «анкилозирующий спондилит» (согласно модифицированным критериям Нью-Йоркской классификации (1984 г.),) установленным не менее чем за три месяца до подписания информированного согласия. Заболевание должно было быть в активной форме (согласно индексу BASDAI и оценке боли в спине: четыре и более балла). Четкие критерии включения пациентов позволяют убедиться, что лекарство получали именно те люди, для которых оно предназначено.
Критерии исключения пациентов: Пациенты с тотальным анкилозом позвоночника (то есть пациенты, у которых все позвонки срослись друг с другом) и с серьезными сопутствующими соматическими или инфекционными заболеваниями вроде ВИЧ-инфекции, гепатита В, С, сифилиса и туберкулеза, или которые которые принимают препараты, способные исказить эффект BCD-085. Четкие критерии исключения пациентов позволяют убедиться, что лекарство не будут получать люди, которым оно точно не поможет.
Какие процедуры использовали: И препарат, и плацебо вводились пациентам в виде инъекций — причем и исследуемый, и контрольный препарат поступали в больницу в одинаковой упаковке. Это обеспечивает надежное заслепление: ни пациенты, ни врачи не смогут отличить контроль от опыта.
Этапы клинических исследований
Клинические исследования — многолетний процесс, который разбивают на разные этапы (иногда этапы называют фазами). Фазы клинических испытаний отличаются по целям и задачам, причем количество участников и время, отведенное на этап клинического исследования, увеличивается от фазы к фазе. Это необходимо, чтобы обеспечить безопасность — ведь эффективный препарат может обладать очень серьезными побочными эффектами, так что вред от его применения будет превышать пользу.
Выделяют четыре фазы клинических испытаний, из которых три являются обязательными для того, чтобы новый препарат в принципе разрешили применять у человека (то есть зарегистрировали, произвели и отправили в клиники или в аптеки).
Фаза I. Цель этапа — убедиться, что препарат можно применять у людей. Ведь даже при успешном завершении доклинических исследований на животных невозможно точно предугадать, какой эффект новое средство окажет на человека. На этом же этапе дополнительно оценивают фармакокинетические и фармакодинамические свойства лекарственного средства — то есть пытаются понять, как лекарство ведет себя в человеческом организме, и насколько оно безопасно.
Как правило, на первой фазе испытаний препарат применяется впервые. Самыми первыми препарат «пробуют» люди из небольшой группы здоровых добровольцев. Обычно это 20-200, но в некоторых случаях — всего 15-30 человек. Пациенты с реальным заболеванием в I фазе испытаний участвуют реже. Первая фаза испытаний длится несколько месяцев.
Фаза II. Цель этапа — дополнительно оценить безопасность лекарства и выяснить, работает ли препарат. На этой фазе иногда выбирают схему лечения или уточняют терапевтическую дозу исследуемого препарата — на этот раз с точки зрения его эффективности.
Во II фазе испытаний участвуют до 300 пациентов с заболеванием, для лечения которого предполагается применять исследуемый препарат. Длится вторая фаза от нескольких месяцев до двух лет.
Результаты II фазы клинических испытаний позволяют сделать только предварительные выводы об эффективности лекарства. Чтобы сделать окончательный вывод об эффективности и безопасности нового лекарства, нужно больше участников и времени.
Пример от BIOCAD
Результаты клинического испытания: препарат Эфлейра®
Препарат успешно прошел I, II и III фазы клинических испытаний. В нашей стране лекарство зарегистрировано не только как препарат против спондилита, но и в качестве средства лечения среднетяжелого и тяжелого псориаза и псориатического артрита. Два последних показания подтвердили результаты еще двух клинических исследований II и III фазы: BCD-085-2 и BCD-085-7 (PLANETA).
Фаза III. Цель этапа — сравнить новый препарат со стандартным лечением. На этой фазе уже используют двойные слепые испытания и оценивают вероятность дальнейшей регистрации препарата.
В III фазе клинических испытаний участвует 300-3000 пациентов с заболеванием, для лечения которого предполагается применять исследуемый препарат. Длится фаза в среднем от года до четырех лет.
Если препарат подтверждает эффективность, установленную на II этапе, а выявленные побочные эффекты оказываются переносимыми, разработчики препарата начинают собирать документы для его регистрации.
Фаза IV называется постмаркетинговой — этот этап клинического испытания начинается, когда готовое лекарство уже поступает в продажу. Часто такие клинические рекомендации проводятся «независимо», то есть такие испытания не финансируются фармацевтическими компаниями. В случае с процедурой условной регистрации, а также положением 61-ФЗ, четвертой фазой могут выступать КИ, проведение которых обязательно по заказу разработчика. Это очень важный этап, особенно если речь идет о препарате, который прошел процедуру условной регистрации. Но даже если лекарство зарегистрировали «по всем правилам», IV фаза все равно важна — именно она позволяет выявить краткосрочные и долгосрочные побочные эффекты и редкие противопоказания.
В IV фазе клинических испытаний обычно участвуют тысячи пациентов. Длятся такие испытания много лет. Иногда их продолжат проводить, даже если препарат давным-давно вошел в клиническую практику. Чаще всего это происходит в ситуации, когда «старое» лекарство планируют использовать по новым показаниям. Иногда в результате таких испытаний «новую жизнь» получают уже забракованные лекарства.
Так произошло и с талидомидом. В 1964 году израильский дерматолог Яков Шескин нашел упаковку уже запрещенного к тому времени препарата, и назначил его пациенту с лепрозной узловатой эритемой — крайне болезненным кожным проявлением лепры (проказы). К удивлению доктора, страдавший от бессонницы пациент наконец-то смог проспать 20 часов, а потом впервые за долгое время поднялся с постели без посторонней помощи. В 1998 году FDA зарегистрировала талидомид в качестве средства от лепрозной узловатой эритемы.
Этика клинических испытаний: информированное согласие
Мы уже писали, что в клиническом испытании препаратов принимают участие только пациенты или добровольцы, подписавшие информированное согласие на участие в исследовании. В основе информированного согласия, которое подписывает каждый пациент, лежат этические принципы, отраженные в Хельсинкской Декларации Всемирной Медицинской Ассоциации (ВМА).
Хельсинкская Декларация гласит: «При проведении любого исследования с участием людей в качестве субъектов каждый потенциальный субъект исследования должен быть надлежащим образом проинформирован о целях, методах, ожидаемой пользе и возможном риске исследования, а также о неудобствах, которые могут быть вызваны экспериментом. Участники исследования должны быть проинформированы о том, что они имеют неограниченное право отказаться от участия в исследовании и в любое время взять назад согласие на участие. Врач должен получить такое согласие — свободное и информированное — от субъекта исследования, желательно в письменном виде».
В реальной международной практике «устное» согласие пациента на эксперимент давно недопустимо. Информированное согласие пациента обязательно нужно задокументировать — с помощью подписанной и датированной формы Информированного Согласия (ИС), которое описано в стандарте GCP (ICH Harmonized Tripartite Guideline for GCP).
Фактически, ИС — это упрощенное описание/содержание протокола исследования, в который добавили важную информацию, без которой согласие нельзя считать по-настоящему «информированным». В нем описаны цели эксперимента, процедуры, которым будут подвергать участников, предполагаемые риски и польза, время эксперимента, условия возмещения возможных расходов и вознаграждения за участие (если, конечно, это предусмотрено). Само собой, пациентам сообщают, что у них есть вероятность попасть в группу контроля — и в принципе не получить никакого лекарства (в случае плацебо), или получить стандартную терапию при исследуемом заболевании.
Очень важно, чтобы пациенты подписали ИС до начала любых действий, которые предусматривает клиническое испытание — если, конечно, с этого действия не начинается любое стандартное обследование. О любых изменениях в порядке проведения испытаний пациента тоже обязаны предупредить. Сделать это нужно своевременно — то есть за некоторое время до того, как изменения вступят в силу.
Что такое этический комитет, и зачем он нужен?
Информированное согласие должна одобрить не только компания-производитель исследуемого лекарства, но и официальные органы страны, в которой проходит клиническое исследование. Проверять, насколько правильно составлено ИС, должны члены этического комитета (ЭК).
К сожалению, в нашей стране пока не существует единого этического комитета, который бы полностью отвечал международным нормативным требованиям и в который могли бы обращаться все компании, которые занимаются разработкой лекарств. Если бы такой орган появился, это бы могло ускорить начало и старт клинических испытаний.
В России за соблюдение этики клинических испытаний отвечают локальные клинические комитеты (ЛЭК-и). Один из таких комитетов действует при Сеченовском Университете.
«Сеченовский» ЛЭК создали еще в 1993 году. Его задача — обеспечение прав, безопасности и благополучия добровольцев и пациентов, которые участвуют в клинических испытаниях, организованных на базе Сеченовки. Без разрешения этой организации отдел доклинических и клинических испытаний не имеет права начать клинические испытания препарата, предоставленного фармкомпанией-заказчиком.
Кто и как контролирует проведение клинических испытаний
Контроль со стороны фармкомпаний. «Над тем, чтобы клинические испытания прошли с соблюдением всех требований, в фармацевтической компании работают две структуры: департамент клинических исследований и департамент клинической разработки, — рассказывает Екатерина Докукина. — Сотрудники этих департаментов следят за соблюдением всех нормативных требований и рекомендаций, разрабатывают и готовят документацию о клиническом исследовании, отвечающую всем необходимым требованиям. В их задачу входит своевременный сбор и предоставление качественных и достоверных данных, которые нужны для регистрационного досье».
Пример от BIOCAD
Как контролируют клинические испытания препарата Эфлейра® Сотрудники компании BIOCAD контролируют качество проведения исследования на двух уровнях:
- регулярно проверяют (мониторируют) правильность составления и заполнения медицинской документации
- проверяют качество полученных данных
На всех этапах предусмотрены независимые аудиты клинических центров и документации исследования. Независимый аудит подтверждает, что сотрудники клиник, в которых проводятся исследования, точно выполняют все требования, которые были предусмотрены в протоколе клинического испытания.
Контроль со стороны государства. Если фармацевтическая компания предоставит недостоверные данные о результатах клинических исследований, это может стоить здоровья (а порой и жизни) пациентов, которые будут принимать новое лекарство. Допуская лекарственный препарат на рынок и выдавая на него регистрационное удостоверение, государственный орган гарантирует, что данный препарат не только безопасен, но и эффективен. Поэтому, прежде чем зарегистрировать лекарство, государственные регуляторы стремятся убедиться, что клинические испытания проведены по определенным правилам.
Эти правила называются стандартами GCP (от «Good Clinical Practice» — надлежащая клиническая практика). Они описывают порядок проведения клинических исследований лекарственных средств — это международные правила планирования и проведения исследований с участием пациентов или здоровых добровольцев (в терминологии стандарта — субъектов исследования). Соблюдение стандарта GCP гарантирует соблюдение прав субъектов исследования и качество, и достоверность полученных в результате исследования данных.
В нашей стране требования к клиническим исследованиям закреплены в положениях нормативных актов, регламентирующих проведение указанных исследований — в частности, 7 глава Федерального закона от 12.04.2010 №61-ФЗ «Об обращении лекарственных средств». Кроме того, в России применяются и нормативные акты ЕАЭС, в частности правила надлежащей клинической практики Евразийского экономического союза, утв. Решением Совета ЕЭК от 03.11.2016 N 79.
Будущее клинических испытаний
«Вероятно, в дальнейшем все больше внимания будет уделяться изучению социально-значимых параметров, таких как качество жизни и его изменение на фоне терапии, — рассказывает директор департамента клинических испытаний компании BIOCAD Екатерина Докукина, — Это нужно не только для того, чтобы учитывать большее число параметров, связанных с самочувствием пациентов, но и увеличит вес этих данных при интерпретации результатов клинического исследования».
Источник


