Испытание лекарств методом «ускоренного старения»
 Промышленное производство лекарств стремительно увеличивается. Современные требования к их качеству постоянно ужесточаются и заставляют проводить новые научные эксперименты, помогающие выявить сроки годности и стабильность препаратов. Самым эффективным опытом по-прежнему остается процесс разложения веществ или «метод ускоренного старения», воздействующий на лекарства влагой, светом, температурой, pH и т. д.
Промышленное производство лекарств стремительно увеличивается. Современные требования к их качеству постоянно ужесточаются и заставляют проводить новые научные эксперименты, помогающие выявить сроки годности и стабильность препаратов. Самым эффективным опытом по-прежнему остается процесс разложения веществ или «метод ускоренного старения», воздействующий на лекарства влагой, светом, температурой, pH и т. д.
Условия работы
Для исследований по проверке препаратов применяются климатические камеры, термические шкафы и другое оборудование, способное обеспечивать точную запрограммированную температуру в течение необходимого времени. Изучение образцов для определения их срока годности проводится в соответствии с научно-технической документацией. Методика «ускоренного старения» подразумевает высокую температуру, не превышающую предела, который разрушает тару или меняет исходное состояние вещества. Установленный предельный режим без воздействия света в климатической камере и прочих агрегатах для пилюль, таблеток, жидкостей и порошков составляет 60°С, для мазей и линементов — 40°С, а для аэрозолей и суппозиториев — 30°С. Исключение составляют термически устойчивые вещества. Описанные работы могут выполнять только компании, создающие или проектирующие лекарственные средства.
Срок годности по данной технологии выявляется в трех (реже двух) партиях препарата. Экспериментальное хранение проводится под воздействием высоких температур (больше нормы на 10°С). Параметры качества лекарств в этом случае устанавливаются в течение временного отрезка, эквивалентного полугоду его хранения в обычных условиях. Наблюдения в процессе опытов могут производиться только по предписаниям НТД. Если же из-за предусмотренных в документации норм нельзя зафиксировать изменения лекарственных средств, допускаются дополнительные опыты. В заключение нужно сказать, что исследование методом «ускоренного старения» подразумевает размещение образцов в запаянные стеклянные ампулы или трубки в необходимом количестве.
Источник
Метод ускоренного старения лекарственных средств это

Продукты органического синтеза составляют значительную часть арсенала лекарственных средств современной медицины. Несмотря на значительное число лекарственных средств, использующихся в медицинской практике, а это более 20 тысяч наименований, постоянно ведется поиск более эффективных и безопасных. В значительной степени это относится к группам лекарственных средств, использующихся для лечения сердечно-сосудистых и онкологических заболеваний [7]. В этом плане представляют интерес научные исследования, проводимые в Пермской государственной фармацевтической академии [4], направленные на поиск потенциальных лекарственных средств для лечения сердечно-сосудистых заболеваний. В частности, одно из полученных соединений (2-метиланилид N,N-диэтиламиноэтановой кислоты нитрат) на стадии скрининговых исследований показало выраженное антиаритмическое действие. Так, в сравнении со структурным аналогом лидокаином, антиаритмический индекс нового биологически активного соединения (БАС) в 9 раз выше при меньшей в 1,6 раза токсичности.
Исследование нового БАС в качестве потенциального лекарственного средства помимо разработки эффективных способов оценки качества на стадии доклинических испытаний включает установление стабильности и сроков годности.
Изучение вышеуказанных нормативных требований необходимо для установления времени, в течение которого вещество сохраняет неизменными физические, химические, биологические свойства, т.е. удовлетворяет всем требованиям нормативной документации. Обоснование установленного срока годности субстанции включается в раздел регистрационного досье, касающегося способов оценки качества.
Целью настоящего исследования является установление срока годности и стабильности БАС из группы анилидов замещенных карбоновых кислот – потенциального лекарственного средства с выраженным антиаритмическим действием.
Материалы и методы исследования
Субстанция мономекаина – 2-метиланилид-N,N-диэтиламиноэтановой кислоты нитрат. Эксперименты проведены на 5 сериях БАС, полученных в лабораторных условиях по методике [1]. Все образцы БАС предварительно проанализированы в соответствии с требованиями проекта ФС, разработанного автором.
ИК спектры получены на ИК Фурье спектрометре ALPHA-T в виде диска с калия бромидом в соотношении 1:200 мг.
Температура плавления соединения определена на приборе SMP3 (BarIowor Id Scientific).
УФ спектры получены на спектрометре PerkinElmerLambda 45.
Использованы титрованные растворы, реактивы, растворители, индикаторы, соответствующие требованиям ГФ XII изд. [5]. Потенциометрическое титрование проводили с помощью автоматического титратора Titrolineeasy, снабженного магнитной мешалкой и комбинированным электродом. Методики испытания на подлинность и количественного определения, разработаны ранее авторами [2].
Результаты исследования и их обсуждение
При установлении сроков годности руководствовались требованиями ОФС 42-0075-07 ГФ XII [5] и Временной инструкции И-42-2-82 [3]. Согласно указанным документам установление стабильности возможно с использованием следующих способов:
— испытание в условиях долгосрочного хранения;
— испытание в условиях «ускоренного старения» согласно Временной инструкции И 42-2-82.
Метод «ускоренного старения», основанный на законе Вант-Гоффа, устанавливает зависимость между сроком годности вещества и температурой хранения экспериментальной серии субстанции: 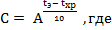
tэ – температура экспериментального хранения,
tхр – температура хранения,
А – температурный коэффициент скорости химической реакции при увеличении температуры на 10 ºС (принят равным 2) [6].
Исследования проводили на 3-х сериях субстанции мономекаина (050713, 080813, 170314) при температуре экспериментального хранения, равной 60 ºС. Образцы помещали в склянки темного стекла с притертыми пробками. Контроль качества проводили через временные промежутки (11,5 дней), эквивалентные 6 месяцам хранения в естественных условиях по показателям, приведенным в табл. 1.
Нормы качества субстанции мономекаина, контролируемые при установлении срока годности
Источник
Методические указания для самостоятельной работы аспирантов по специальности Фармацевтическая химия, фармакогнозия
ПРОЕКТ
МИНИСТЕРСТВО ЗДРАВООХРАНЕНИЯ РОССИЙСКОЙ ФЕДЕРАЦИИ
ГОСУДАРСТВЕННЫЙ СТАНДАРТ КАЧЕСТВА ЛЕКАРСТВЕННОГО СРЕДСТВА
ОБЩАЯ ФАРМАКОПЕЙНАЯ СТАТЬЯ
Сроки годности ОФС
лекарственных средств Взамен ГФ ХII, ч.1, ОФС 42-0075-07
Требования данной статьи распространяются на методы определения стабильности лекарственных средств, которые лежат в основе установления их сроков годности.
Основные термины и определения
Срок годности – период времени, в течение которого лекарственное средство полностью отвечает всем требованиям нормативной документации, в соответствии с которой оно было выпущено и хранилось.
Стабильность — способность лекарственного средства сохранять химические, физические, микробиологические и биофармацевтические свойства в определенных границах на протяжении срока годности.
Долгосрочные испытания стабильности – испытания, проводимые в соответствии с заявленными в нормативной документации условиями хранения лекарственного средства с целью установления или подтверждения срока годности.
Испытания стабильности методом «ускоренного старения» – испытания, проводимые при повышенной температуре с целью установления или подтверждения срока годности лекарственного средства.
Стресс-исследования – испытания стабильности в стресс-условиях, проводимые с целью исследования вынужденного процесса разложения (установления продуктов и механизмов разложения) лекарственного средства.
Матричный метод исследования стабильности (matrixing) – метод исследования, при котором в определенный момент времени исследуется лишь подгруппа из общего числа образцов всех комбинаций факторов, подлежащих изучению.
Метод крайних вариантов (bracketing) – метод изучения стабильности, при котором во всех временных точках по полному протоколу тестируют только образцы с крайними вариантами факторов.
Экстраполяция − способ получения информации о будущих данных на основании имеющихся.
Дата производства/изготовления – дата завершения последней стадии производства/изготовления лекарственного средства.
Дата выпуска – дата поступления или разрешения поступления лекарственного средства в обращение.
Общие положения
Срок годности лекарственного средства устанавливается экспериментально при хранении в течение определенного времени в условиях и упаковке, регламентируемых нормативной документацией, и по мере накопления данных он может быть изменен как в сторону увеличения, так и в сторону уменьшения.
В основу определения сроков годности положено изучение стабильности лекарственного средства с использованием химических и физико-химических методов анализа, указанных в общих фармакопейных статьях, а также, в случае необходимости, других специальных методов исследований, например, биологических методов анализа, фармакологических испытаний.
Срок годности лекарственных препаратов устанавливают независимо от сроков годности фармацевтических субстанций. Однако для лекарственных препаратов, произведенных путем фасовки фармацевтических субстанций, следует учитывать, что стабильность лекарственных препаратов может зависеть от остаточного срока годности используемой фармацевтической субстанции.
Также необходимо учитывать и оценивать влияние на стабильность лекарственных препаратов фактора длительного хранения нерасфасованной и промежуточной продукции до передачи с одного производственного участка на другой или на участок упаковки.
Первоначальный срок годности лекарственного средства, который, как правило, должен быть не менее двух лет, определяет производитель (разработчик) лекарственного средства при подготовке проекта нормативной документации.
После регистрации лекарственного средства и начала промышленного выпуска производитель (разработчик) обязан продолжить работы по изучению стабильности лекарственного средства с целью подтверждения или уточнения его срока годности.
Не рекомендуется устанавливать сроки годности более 5 лет, даже если результаты изучения стабильности позволяют это сделать.
Изменение сроков годности лекарственных средств или условий хранения утверждается в установленном порядке на основе данных, подтверждающих обоснованность заявленных изменений.
Начальной датой отсчета срока годности лекарственного средства является дата его производства/изготовления.
На основании изучения свойств лекарственного средства устанавливают оптимальные требования к первичной и вторичной упаковке и условиям хранения.
После установления оптимальных требований к первичной и вторичной упаковке и условиям хранения производитель (разработчик) лекарственного средства экспериментально определяет сроки годности лекарственного средства, осуществляя его хранение в рекомендованной упаковке и в указанных условиях с целью обнаружения скрытых факторов, которые могут повлиять на устойчивость лекарственного средства при хранении. Для этого от каждой из не менее чем трех серий образца лекарственного средства, специально произведенных по условиям лабораторного или опытно-промышленного регламента, отбирают и укупоривают часть из них в количестве, достаточном для исследования стабильности лекарственного средства.
При изучении стабильности лекарственных средств, расфасованных в крупногабаритную первичную упаковку (фляги, бутыли, железные бочки, жестяные барабаны, пакеты полимерные, мешки бумажные трех- и четырехслойные и т.д.), допускается использование аналогичной упаковки меньшей емкости, достаточной для моделирования условий оригинальной упаковки.
Перед началом испытания проводят анализ лекарственного средства по всем показателям, предусмотренным проектом нормативной документации. Испытания по показателям, которые не изменяются в процессе хранения, а также испытания по показателям, изменения которых в процессе хранения не происходят в сторону ухудшения качества лекарственного средства, допускается не включать в протоколы исследования стабильности лекарственного средства.
На основании полученных результатов производитель (разработчик), изучающий стабильность лекарственного средства, определяет срок годности с указанием вида упаковки, требуемых условий хранения и транспортирования и вносит эти данные в проект нормативной документации.
При исследовании стабильности лекарственного препарата одновременно с изучением стабильности действующего и вспомогательного веществ оценивают их совместимость.
Для некоторых лекарственных препаратов в таких лекарственных формах, как растворы, суспензии, эмульсии и др. при необходимости проводят исследования по изучению влияния на их стабильность отрицательных температур (циклы замораживания и оттаивания).
Долгосрочные исследования стабильности лекарственных средств
Долгосрочные испытания должны проводиться в рекомендованной для данного лекарственного средства первичной и вторичной упаковке при постоянной верхней (наиболее высокой) температуре установленного режима хранения в течение всего предполагаемого срока годности.
В ряде случаев могут требоваться дополнительные испытания при нижней температуре установленного режима хранения (например, для мягких лекарственных форм, для которых возможны изменения их физико-химического состояния при пониженных температурах).
Образцы лекарственных средств, находящиеся на изучении их стабильности, подлежат проверке по показателям качества нормативной документации в следующие сроки:
— в течение первого года хранения – через каждые 3 месяца;
— в течение второго и третьего года хранения – через каждые 6 месяцев;
— после третьего года хранения – через каждые 12 месяцев.
Долгосрочные исследования стабильности лекарственных средств являются обязательным испытанием для установления их срока годности.
Испытания методом «ускоренного старения»
Метод «ускоренного старения» преимущественно используется для определения сроков годности фармацевтических субстанций, представляющих собой вещества с установленным химическим строением, и лекарственных препаратов, содержащих эти вещества в качестве действующих.
Срок годности, установленный с помощью метода «ускоренного старения», не должен превышать трех лет, для антибиотиков, полученных микробиологическим или полусинтетическим путем, и их лекарственных форм – двух лет. Метод не применим для увеличения ранее установленного срока годности лекарственного средства на период свыше трех лет.
Метод «ускоренного старения» заключается в выдерживании испытуемого лекарственного средства при температурах, превышающих температуру его хранения. При повышенных температурах, как правило, ускоряются протекающие в лекарственных средствах физико-химические процессы, приводящие со временем к нежелательным изменениям качества. Таким образом, при повышенной температуре промежуток времени, в течение которого контролируемые показатели качества лекарственного средства сохраняются в допустимых пределах (экспериментальный срок годности), искусственно сокращается в сравнении со сроком годности при температуре хранения. Это позволяет значительно сократить время, необходимое для установления срока годности.
По результатам, полученным в процессе «ускоренного старения» лекарственного средства, можно решить также обратную задачу, т.е. установить температуру хранения, обеспечивающую какой-либо заданный срок годности.
Срок годности С при температуре хранения tхр. связан с экспериментальным сроком годности СЭ при повышенной температуре экспериментального хранения tэ следующей зависимостью:
где коэффициент соответствия
Температурный коэффициент скорости химической реакции A принят равным 2,5.
Примечания. 1. Приведенная зависимость основана на правиле Вант-Гоффа о 2-4-кратном росте скоростей химических реакций при увеличении температуры на 10°С.
2. В отдельных случаях возможно использование экспериментально определенных уточненных значений коэффициента A, а также прогнозирования сроков годности на основании более строгих зависимостей, например уравнения Аррениуса.
В таблице 1 приведены значения коэффициентов соответствия K для различных значений разности температур экспериментального и обычного хранения при A = 2,5.
Таблица 1 – Значения коэффициентов соответствия K
Для опытов по «ускоренному старению» лекарственных средств должны использоваться термостаты, термошкафы, климатические камеры или другие устройства, позволяющие автоматически поддерживать заданную температуру экспериментального хранения tэ в течение всего опыта с точностью ±2 °С.
Наиболее высокая температура экспериментального хранения должна обеспечивать получение результатов, необходимых для оценки сроков годности в кратчайшие промежутки времени. Однако эта температура не должна превышать пределов, за которыми происходят изменения агрегатного состояния лекарственного средства или разрушение упаковочного материала.
Рекомендуются следующие предельные температуры экспериментального хранения:
— для индивидуальных веществ : + 60 °С;
— для таблеток, капсул, парентеральных растворов в стеклянной упаковке: + 60 °С;
— для мазей, линиментов и парентеральных растворов в полимерной упаковке: + 40 °С;
— для суппозиториев и аэрозолей: + 30 °С.
Воздействие света на испытуемые образцы должно быть исключено.
Не рекомендуется установление срока годности методом «ускоренного старения» для эмульсий.
Определение сроков годности методом «ускоренного старения» должно проводиться на не менее чем трех сериях лекарственного средства.
Примечание. Рекомендуется проводить изучение стабильности лекарственных препаратов, приготовленных из разных серий фармацевтической субстанции.
Температура экспериментального хранения (tэ) должна превышать температуру хранения (tхр) не менее, чем на десять градусов.
Наблюдение за качеством изучаемых образцов лекарственного средства должно проводиться по показателям, предусмотренным нормативной документацией с учетом общих положений настоящей статьи.
Показатели качества лекарственного средства в процессе «ускоренного старения» определяют через промежутки времени, эквивалентные шести месяцам хранения при условиях хранения, указанных в проекте нормативной документации.
Количество образцов лекарственного средства, предназначенных для экспериментального хранения, должно быть достаточным для проведения исследований, предусмотренных планом эксперимента.
Началом экспериментального хранения считается момент помещения лекарственного средства в термостатирующее устройство, а концом его – либо момент, когда истекает экспериментальный срок хранения, соответствующий не менее чем двухлетнему сроку годности, либо момент, когда показатели качества лекарственного средства перестают удовлетворять требованиям нормативной документации.
Сроки экспериментального хранения при различных температурах представлены в таблице 2.
Таблица 2 – Сроки экспериментального хранения
Сроки экспериментального хранения, сутки
Примечание. * — в случае подтверждения срока годности, равного ранее утвержденному
Для вычисления срока годности экспериментальный срок годности, выраженный в сутках (или часах), умножают на коэффициент соответствия K (см. таблицу 1).
Если промежуток времени Со между датой производства/изготовления лекарственного средства и началом его экспериментального хранения превышает 30 суток (но не более 90 суток), и оно в это время хранилось в обычных условиях, расчет срока годности С проводят по уравнению C=K×Cэ+Cо.
Если сроки годности, установленные на различных сериях лекарственных средств, отличаются друг от друга, за срок годности принимают минимальное из полученных значений.
При необходимости tхр.,позволяющую обеспечить заданный срок годности С, рассчитывают по формуле:
Метод экстраполяции
При достаточном обосновании допускается экстраполяция данных, полученных по результатам долгосрочного хранения.
Экстраполяцию проводят с помощью статистической обработки данных. Если полученные результаты свидетельствуют о незначительной деградации и малой вариации, статистический анализ может не проводиться.
Методом экстраполяции предлагаемый срок годности может быть увеличен не более чем в два раза, но не более чем на 12 месяцев по сравнению с долгосрочными испытаниями.
Данные, полученные с использованием метода экстраполяции, должны быть подкреплены обязательствами предприятия (разработчика) по продолжению изучения стабильности в условиях долгосрочных испытаний в течение всего заявленного срока годности.
Метод крайних вариантов
При изучении стабильности лекарственных средств допускается проведение исследования крайних вариантов.
При использовании метода крайних вариантов во всех временных точках по полному протоколу тестируют только образцы с крайними (предельными) вариантами факторов (например, дозировки, размер контейнера и (или) номинальный объем). Такой протокол предполагает, что стабильность любых промежуточных вариантов соответствует стабильности исследуемых крайних вариантов.
Исследование крайних вариантов допускают в отношении нескольких дозировок с пропорциональным составом; в случае одного и того же вида упаковки, если при прочих равных условиях имеются различия в размере упаковки или номинальном объеме лекарственного препарата.
Матричный метод
При использовании матричного метода в определенный момент времени исследуется лишь подгруппа из общего числа образцов всех комбинаций факторов, подлежащих изучению. В очередной момент времени проводят исследование другой подгруппы образцов всех комбинаций факторов. К различным факторам одного и того же лекарственного препарата, например, относят совокупность различных серий, различных дозировок, различных размеров одной и той же упаковки и, в ряде случаев, различные укупорочные системы одной упаковки.
Исследование влияния упаковки на стабильность
лекарственного средства
Изменения качества лекарственного препарата могут быть вызваны взаимодействием лекарственного средства и системы упаковки, включающей укупорочные средства. Если для жидких лекарственных препаратов (кроме тех, что находятся в запаянных ампулах) нельзя исключить отсутствие взаимодействия, то в испытания на стабильность включают образцы в перевернутом или горизонтальном положениях (т.е. образцы, которые контактируют с укупорочным средством, например, пробкой) наряду с вертикально установленными образцами для определения влияния материала укупорочного средства (пробки) на качество лекарственного препарата. Результаты экспериментальных исследований должны фиксировать все сочетания различных систем упаковки (укупорки), анализируемых лекарственных средств.
Для лекарственных средств в многодозовой упаковке кроме стандартных данных, необходимых для традиционной упаковки одноразового использования (например, флакона), заявитель должен провести испытания, подтверждающие их способность выдержать условия повторяемого открывания и закрывания и при этом сохранение качества и эффективности лекарственного средства на протяжении всего срока применения.
К основным факторам, оказывающим влияние на лекарственный препарат после вскрытия упаковки, относятся: микробное загрязнение и физико-химическая деградация.
В исследование стабильности лекарственных препаратов после вскрытия первичной упаковки следует включать не менее двух серий; при этом, по крайней мере, одна серия должна быть с истекающим сроком годности.
Проверку показателей на соответствие требованиям нормативной документации осуществляют в первую временную точку, как минимум одну промежуточную, а также в последнюю временную точку предлагаемого срока годности вскрытого лекарственного препарата.
Анализ лекарственного препарата проводят по всем показателям нормативной документации, которые могут меняться в процессе хранения (за исключением показателей, изменения по которым в процессе хранения не могут происходить в сторону ухудшения качества), и обязательно должны включать контроль на микробиологическую чистоту или стерильность.
Исследования стабильности лекарственных препаратов после восстановления или разведения
Если инструкцией по медицинскому применению предполагается возможность хранения восстановленного твердого лекарственного препарата или разведенного концентрированного лекарственного препарата в течение определенного периода времени, должны проводиться исследования стабильности приготовленного таким образом препарата.
Цель изучения стабильности восстановленных препаратов – определить срок, в течение которого после восстановления или разведения лекарственного препарата его качество продолжит соответствовать требованиям нормативной документации и лекарственный препарат может применяться по назначению.
Изучению стабильности подлежат восстановленные лекарственные препараты, приготовленные с использованием всех возможных для растворения/разведения лекарственных препаратов растворителей, указанных в инструкции по медицинскому применению.
Условия хранения восстановленного лекарственного препарата могут отличаться от условий хранения исходного лекарственного препарата.
Для подтверждения стабильности восстановленного лекарственного препарата допускается предоставлять данные, полученные для двух серий.
Проверку показателей на соответствие требованиям нормативной документации рекомендуется осуществлять в первую и последнюю временные точки предлагаемого срока годности восстановленного лекарственного препарата.
Анализ лекарственного препарата проводят по всем показателям, которые могут меняться в процессе хранения, и он обязательно должен включать контроль на стерильность или микробиологическую чистоту.
Стресс-исследования и фотостабильность
Помимо установления срока годности и выбора условий хранения изучение стабильности оригинальных лекарственных препаратов и фармацевтических субстанций проводится с целью установления наиболее вредного влияния внешних факторов (высокие или низкие температуры, влага, кислород и другие компоненты воздуха, свет и т.п.) в зависимости от времени и условий их воздействия.
Стресс-исследования допускается проводить на одной серии лекарственного средства. Неотъемлемой частью стресс-исследований является исследование фотостабильности. Объем исследований лекарственного средства должен определяться на основании наличия или отсутствия изменений, возникших в результате влияния света.
Вопросы по теме для самостоятельного изучения (для зачета, экзамена), темы рефератов.
1. Изменения, происходящие при хранении лекарственных средств. Реакции окисления, гидролиза, сольволиза.
2. Влияние внешних факторов на стабильность лекарственных средств.
3. Внутренняя перегруппировка в структуре лекарственных средств.
4. Методы исследования структуры лекарственных средств при воздействии физико – химических факторов.
5. Влияние микробной обсемененности на стабильность лекарственных средств.
Список литературы.
1. Фармацевтическая химия: учебное пособие [Электронный ресурс] / Аксенова Э.Н., Андрианова О.П., Арзамасцев А.П. и др.; под ред. А.П. Арзамасцева. / 2-е изд., испр. — М.: ГЭОТАР-Медиа, 2008. — 640 с. – Режим доступа: http://www.studmedlib.ru/ru/book/ISBN9785970407448.html
2. ЭБС «Консультант плюс» — разработка правовых систем http://www.consultan.ru
МЕТОДИЧЕСКИЕ УКАЗАНИЯ ДЛЯ САМОСТОЯТЕЛЬНОЙ ВНЕАУДИТОРНОЙ РАБОТЫ ОБУЧАЮЩИХСЯ (ТЕМА 2 – 3)
Тема: Анализ лекарственных веществ в биологических жидкостях. Основные типы химических превращений лекарственных веществ в организме. Связь между концентрацией лекарственного вещества и его действием. Особенности качественного и количественного анализа лекарственных веществ и их метаболитов в биологических жидкостях.
Фармакокинетика как основа для разработки методов индивидуализации и оптимизации лекарственных средств. Термины и определения. Методы исследования. Роль физико-химических методов анализа лекарственных ве-ществ в фармакокинетических исследованиях. Требования, предъявляемые к методам анализа лекарственных веществ при изучении биологической доступности и фармакокинетики. Общая характеристика оптических, хроматографических и других физико-химических методов применительно к проблеме.
ИНФОРМАЦИОННЫЙ БЛОК
Биофармацевтический анализ — новое перспективное направление фармацевтической химии. Задачей биофармацевтического анализа является разработка способов выделения, очистки, идентификации и количественного определения ЛВ и их метаболитов в таких биологических жидкостях, как моча, слюна, кровь, плазма или сыворотка крови и др. Только на основе применения таких методик можно выполнять биофармацевтические исследования, т.е. изучать вопросы всасывания, транспорта и выведения ЛВ, его биологическую доступность, процессы метаболизма. Все это позволяет предупреждать возможное токсическое воздействие ЛС, разрабатывать оптимальные режимы фармакотерапии и контролировать процесс лечения. Особенно важно определять в биологических жидкостях концентрацию ЛВ, когда они наряду с терапевтическим эффектом проявляют токсичность. Необходимо также контролировать содержание ЛВ в биологических жидкостях больных, страдающих желудочно-кишечными заболеваниями и заболеваниями печени и почек. При таких заболеваниях изменяются процессы всасывания, нарушаются метаболические процессы, замедляется выведение ЛВ из организма.
Биологические жидкости — очень сложные объекты для выполнения анализа. Они представляют собой многокомпонентные смеси, включающие большое число неорганических и органических соединений различной химической структуры: микроэлементы, аминокислоты, полипептиды, белки, ферменты и др. Их концентрация колеблется от 10 мг/мл до нескольких нанограммов. Даже в такой относительно простой физиологической жидкости, как моча, идентифицировано несколько сотен органических соединений. Всякий биологический объект — очень динамичная система. Ее состояние и химический состав зависят от индивидуальных особенностей организма, воздействия факторов внешней среды (состав пищи, физическая и психическая нагрузка и т.д.). Все это еще в большей степени усложняет выполнение биофармацевтического анализа, так как на фоне столь большого количества сложных по химическому строению органических веществ нужно определять нередко очень малые концентрации ЛВ. Вводимые в биологические жидкости ЛВ в процессе биологической трансформации образуют метаболиты, количество которых нередко исчисляется несколькими десятками. Выделение этих веществ из сложных смесей, разделение на индивидуальные компоненты и установление химического состава — задача необычайно трудная.
Таким образом, можно выделить следующие особенности биофармацевтического анализа:
1. Объекты исследования представляют собой многокомпонентные смеси соединений.
2. Количества определяемых веществ, как правило, исчисляются микрограммами и даже нанограммами.
3. Исследуемые ЛВ и их метаболиты находятся в среде, состоящей из большого числа природных соединений (белков, ферментов и др.).
4. Условия выделения, очистки и анализа исследуемых веществ зависят от вида биологической жидкости, подвергаемой
Помимо теоретического значения, которое имеют исследования в области биофармацевтического анализа для изучения вновь создаваемых ЛВ, несомненна и практическая роль этой отрасли знаний.
1. Связь проблем фармацевтической химии с фармакокинетикой и фармакодинамикой
К концу 50-х — началу 60-х гг. XX в. было обращено внимание на зависимость фармакологической активности от таких физических факторов, как степень измельчения и явление полиморфизма, а также от технологических процессов получения ЛС. Возникло своеобразное противоречие между существовавшими нормами оценки качества и фактическим действием ЛС. Последние по результатам аналитического контроля соответствовали в одинаковой степени требованиям фармакопеи (ФС), но различались по фармакологическому эффекту. Так возникло понятие о терапевтической неэквивалентности ЛС. Оно означает, что одни и те же ЛФ, содержащие одинаковые количества ЛВ, но изготовленные разными способами, оказывают неодинаковый терапевтический эффект. Установить причину такого явления можно только проведением биофармацевтических и фармакокинетических исследований.
Сформулированные к настоящему времени основные принципы установления количественных соотношений между химической структурой и фармакологической активностью можно представить в виде трех основных стадий. Первая — биофармацевтическая — включает исследование исходного биологически активного вещества и создание на его основе готовой лекарственной формы. Вторая стадия — фармакокинетическая — включает исследование таких происходящих в организме кинетических процессов, как всасывание, распределение, связывание с белками, биотрансформация и выведение (экскреция) ЛВ. Эти процессы изучаются в сопоставлении с фармакологическим или токсическим действием этих веществ на организм. Третья стадия — фармакодинамическая — включает исследование взаимодействия ЛВ с рецептором и влияние на регуляторные системы. Только на этой стадии в полной мере проявляется и является специфичной взаимосвязь химической структуры ЛВ и его фармакологического эффекта. Следовательно, биологическая активность лекарств объясняется последовательно происходящими тремя фазами: биофармацевтической, фармакокинетической и фармакодинамической.
Изучение механизма качественных и количественных изменений ЛВ в органах и биологических жидкостях организма входит в задачу фармакокинетики. На фармакокинетику Л В оказывают влияние различные факторы: возрастные, генетические, половые, масса тела, питание, беременность, а также различные патологические процессы, например заболевания печени, почек, сердечно-сосудистой системы, желудочно-кишечного тракта, эндокринные, инфекционные и другие заболевания.
Проведение фармакокинетических испытаний осуществляется на стыке нескольких наук и требует участия различных специалистов: врача-клинициста, врача-лаборанта, биохимика, провизора-аналитика, микробиолога, а в ряде более сложных случаев также биофизика, математика, программиста.
Исследования в области фармакокинетики проводятся на животных в период предклинических испытаний, во время клинических испытаний, при разработке технологии производства и контроля качества ЛФ, а также продолжаются после внедрения ЛС в медицинскую практику.
Проведение фармакокинетических исследований возможно только на основе применения современных методов биофармацевтического анализа, позволяющих проследить процесс всасывания и распределения ЛВ в органах и тканях. Они включают выяснение влияния различных биофармацевтических факторов на терапевтическую эффективность Л В; изучение их биологической доступности и разработку методов ее определения; создание способов определения ЛВ и их метаболитов в биологических жидкостях. Основным фармакокинетическим параметром является продолжительность достижения и сохранение максимального уровня концентрации лекарственного вещества в крови, а также скорость и характер ее снижения. Это обусловлено наличием корреляции между терапевтическим эффектом и длительностью циркуляции ЛВ в плазме крови.
Выполнение фармакокинетических исследований ведет к накоплению сведений о количественной оценке ряда кинетических параметров у людей. Так, например, имеются многочисленные данные о связывании большинства применяемых в медицине ЛВ с белками плазмы, биодоступности при приеме внутрь, выделении неизмененного ЛВ с мочой (в процентах к дозе), а также о терапевтических концентрациях в плазме крови (мкг/мл) и периоде полувыведения из плазмы крови (в часах) в норме, при почечной и печеночной недостаточности, у людей различного возраста.
Эти данные дают возможность ориентироваться в понимании механизма действия, прогнозирования химической структуры ЛВ, обусловливающей направленное действие. Иначе говоря, результаты фармакокинетических исследований существенно дополняют данные о связи между химической структурой и фармакологической активностью ЛВ.
Эффективность воздействия ЛВ находится в зависимости от путей его введения в организм.
Процесс поступления лекарственного вещества из места введения в кровь обозначают термином «всасывание». Этот процесс происходит при всех путях введения ЛС, за исключением внутривенного и внутриартериального. Всасывание зависит от пути введения и растворимости ЛВ, а также от кровотока в месте введения. При прохождении через слизистые оболочки всасывание определяется растворимостью в липидах, рН среды в желудке и в кишечнике, ионизацией, активностью их транспорта, скоростью абсорбции в различных отделах желудочно-кишечного тракта. Значительным превращениям подвергаются ЛВ под влиянием ферментов желудочно-кишечного тракта и печени, вызывающих образование различных метаболитов. На скорость и полноту всасывания ЛВ оказывают влияние моторика желудочно-кишечного тракта, объем и состав пищи, интервал времени между едой и приемом ЛС, воздействие пищи на секрецию желудочного сока, объем жидкости, принимаемой вместе с ЛС.
Попав в системный кровоток, ЛВ распределяется по тканям организма. Этот процесс носит название распределения. Он зависит от множества различных факторов, наиболее важными из которых являются растворимость в липидах, степень связывания с белками плазмы крови, интенсивность кровотока. Растворимые в липидах ЛВ быстро распространяются по всему организму. Многие ЛВ в силу большого физико-химического сродства к белкам плазмы крови (особенно к альбумину) связываются ими (иногда на 90%) и ограничивают их концентрацию в тканях в месте действия. Образовавшиеся комплексы с белком лишены фармакологической активности. Наибольшая интенсивность системного кровотока наблюдается в тех органах и тканях, которые активно перфузируются кровью, — в сердце, печени, почках. Значительно медленнее насыщаются ЛВ мышцы, слизистые оболочки, кожа, жировая ткань. Для достижения терапевтической концентрации в этих тканях необходимо нередко несколько часов. Важным фактором, определяющим распределение ЛВ, является также скорость его диффузии в различные ткани.
Таким образом, всасывание и распределение лекарственного вещества зависят не только от путей введения, но и от многих других факторов, обусловленных как физико-химическими свойствами Л В, так и физиологическими процессами, происходящими в организме.
2. Фармакокинетические параметры
Количественно характеризуют процессы, происходящие с ЛВ в организме, основные фармакокинетические параметры, которые отражают связь между концентрацией ЛВ в биологических жидкостях и его фармакологическим действием.
Константа скорости всасывания — параметр, отражающий скорость поступления (ч, мин) ЛВ из места введения в системный кровоток. Используют этот параметр при всех путях введения, кроме внутривенного и внутриартериального.
Константа скорости элиминации (ч, мин-1) характеризует скорость удаления (элиминации) ЛВ из организма путем экскреции или биотрансформации.
Константа скорости экскреции характеризует скорость выделения ЛВ (ч-1, мин-1) с мочой, слюной, калом, молоком или другими экскретами.
Важным фактором, влияющим на терапевтический эффект, является содержание Л В в организме. Оно зависит от продолжительности выведения или элиминации из организма. Показателем элиминации является клиренс (мл/мин). Общий клиренс — это объем плазмы или крови, из которого за единицу времени ЛВ выводится почками, печенью, легкими или биотрансформируется в организме. Параметр, определяющий скорость очищения организма от лекарственного вещества почками, носит название почечный клиренс, а другими путями — внепочечный клиренс. Клиренс в клинической практике используют для расчета терапевтической или поддерживающей дозы ЛВ в крови.
Объем распределения лекарственного вещества — это гипотетический объем жидкостей организма, который необходим для равномерного распределения всего количества ЛВ в той же концентрации, в которой он содержится в плазме крови. Этот показатель находится в зависимости от пола, возраста, общей массы жиров в организме больного. Распределение ЛВ зависит от таких его физико-химических свойств, как растворимость в воде и в липидах, молекулярная масса, полярность, уровень ионизации. Объем распределения используют для расчета дозы ЛВ, необходимой для достижения нужной концентрации его в крови.
О выведении ЛВ из организма судят по периоду пол у вы веден ия, или полуэлиминации. Под ним понимают время, в течение которого происходит снижение на 50% концентрации ЛВ по сравнению с введенным количеством. За один период полуэлиминации из организма выводится 50%, за два периода — 75%, за три периода — 90% ЛВ.
Равновесная концентрация — это состояние, при котором количества вводимого и адсорбирующегося ЛВ равны между собой. Поэтому при равновесной концентрации содержание ЛВ в организме колеблется между максимальными и минимальными его значениями. Это соответствует оптимальному проявлению клинического эффекта.
Период полуабсорбции (полувсасывания) — время (ч, мин), необходимое для всасывания ЛВ из места введения (кроме внутрисосудистого) в системный кровоток половины введенной дозы.
Период полураспределения (ч, мин) — условный параметр, характеризующий распределение ЛВ между центральной камерой (плазма крови) и периферической камерой (органы, ткани).
Площадь под фармакокинетической кривой — площадь фигуры, ограниченной на графике фармакокинетической кривой и осями координат, одна из которых обозначает концентрацию ЛВ в плазме крови (мкг/мл), а другая — время после введения ЛВ (мин).
2.1 Основы фармакодинамики
Разнообразные изменения, которые происходят в организме под влиянием Л В, называются фармакодинамикой.
Первичная фармакологическая реакция сопровождается процессом переноса протонов и электронов с одного вещества на другое. Это осуществляется за счет различных типов химических связей. Наиболее часто встречается ванн-дер-ваальсов тип связи. Такие связи возникают между двумя функциональными группами, одна из которых входит в состав молекулы ЛВ, а другая — в биологическую молекулу. Ван-дер-ваальсовы связи возникают в тех случаях, когда молекулы находятся на близком расстоянии друг от друга, не превышающем 0,2 нм, а энергия связи составляет 0,836-4,18 кДж/моль.
Наиболее важное значение в действии ЛВ имеют водородные связи (-ОН. О-) с энергией-8,4-21 кДж/моль. Водородная связь появляется только в том случае, если атом, участвующий в ее образовании, располагается на расстоянии не более 0,3 нм. Атом водорода может связывать атомы серы, кислорода, азота, галогенов.
Между ионами, имеющими разноименные заряды, возникают ионные связи. Возможности для их образования в организме практически безграничны ввиду наличия большого количества ионов в биологических средах. Энергия ионных связей составляет -21-42 кДж/моль, но длительность их существования в организме очень непродолжительна и не превышает 10
Немалую роль в фармакологических реакциях играет ион-дипольная связь, имеющая энергию порядка -8,4-21 кДж/моль. Такая связь ориентирует молекулы ЛВ относительно соответствующей функциональной группы фермента или рецептора. Возможны также диполь-дипольные связи, участвующие в фиксации ЛВ на функциональной группе рецептора. Их энергия равна -4,2-12,5 кДж/моль.
Наиболее прочной является ковалентная связь. Она образуется между двумя атомами за счет общей пары электронов и имеет энергию 42-627 кДж/моль.
Таким образом, основой первичного взаимодействия между Л В и тканями организма является процесс, сопровождающийся образованием ван-дер-ваальсовых, водородных, ионных, дипольных связей. Предполагается, что ЛВ притягивается рецептором, затем происходит ориентация его молекулы и, наконец, фиксация молекулы на рецепторном поле. Следовательно, специфический ответ клетки органа или организма в целом происходит после адсорбции ЛВ на рецепторе.
Биофармацевтические и фармакокинетические исследования позволяют решить ряд практических задач, например дать рекомендации по изменению физических или химических свойств Л В для повышения их фармакологической активности; обосновать оптимальный выбор биофармацевтических факторов при производстве тех или иных ЛФ. Практическое значение имеют и такие рекомендации, как уточнение показаний и противопоказаний, установление рациональных терапевтических доз и периодичности их приема в течение суток, определение оптимальных путей введения ЛС в организм, разработка научно обоснованных схем лечения тех или иных заболеваний.
3. Понятие о биофармацевтических факторах
ЛС представляют собой сложные химические системы, которые вступают в определенные взаимодействия с биологическими системами организма. На этот процесс существенно влияют самые различные факторы, известные под названием биофармацевтических факторов. Наиболее существенными из них являются полиморфизм, степень дисперсности, физические и химические свойства вспомогательных веществ, используемых при изготовлении лекарственных форм.
Фармакологическое действие кристаллических веществ зависит от образования полиморфных форм. Полиморфизм — способность вещества одной и той же химической структуры кристаллизоваться в различных формах, т. е. изменять свою сингонию в зависимости от термодинамических условий. Одно и то же вещество при соответствующих условиях может образовывать несколько полиморфных структур, отличающихся друг от друга физическими и физико-химическими свойствами. Они могут отличаться по плотности, удельной теплоемкости, проводимости, оптическим и другим константам. Установить наличие таких модификаций можно по растворимости, температуре плавления, а также с помощью физико-химических методов (ИК-, ЯМР-спектроскопия).
Степень дисперсности оказывает большое влияние на процесс всасывания и терапевтическую активность. Как правило, последняя возрастаете уменьшением размера диспергированных частиц ЛВ. Уменьшение в 30 раз (по сравнению с принятым ГФ) размера частиц кислоты ацетилсалициловой усиливает вдвое ее действие на организм. Если подвергнуть очень тонкому измельчению сульфаниламидные препараты, некоторые препараты гормонов, то адекватная терапевтическая активность при их применении достигается вдвое меньшими дозами. В некоторых случаях, например при применении производных нитрофурана, ЛВ следует назначать в виде крупных кристаллов, чтобы уменьшить раздражающее действие на слизистые желудочно-кишечного тракта.
Биофармацевтические исследования очень важны для оценки роли физических и химических свойств вспомогательных веществ, используемых для приготовления ЛФ. Вспомогательные вещества далеко не индифферентны в химическом и фармакологическом отношении. Они могут снижать фармакологическую активность ЛВ, повышать ее и даже изменять характер фармакологического действия под влиянием различных физических и химических процессов.
4. Способы установления биологической доступности лекарственных средств
Биологическая доступность — это степень всасывания JIB из места введения в системный кровоток и скорость, с которой этот процесс происходит. Такое понятие признано ВОЗ.
Терапевтический эффект зависит от того, какая часть введенного JIB попадет в системный кровоток и затем будет доставлена в те ткани или органы, в которых осуществляется его специфическое действие. Этот показатель характеризует биологическую доступность. При внутривенном введении она равна 100%, при всех других способах применения — всегда ниже 100%. Это вызвано тем, что, прежде чем попасть в кровоток, JIB должно пройти целый ряд биологических мембран клеток слизистой желудка, печени, мышц и т.д.
На биодоступность оказывают влияние биофармацевтические факторы, в частности лекарственная форма, пути ее введения, индивидуальные особенности организма человека, физиологическое и патологическое состояние желудочно-кишечного тракта, сердечно-сосудистой системы, печени, почек и др.
Биодоступность изучают путем сравнительного исследования изменений концентраций JIB в плазме крови или в моче после введения испытуемой и стандартной ЛФ. Поскольку внутривенное введение обеспечивает 100%-ную биодоступность, можно установить абсолютную биодоступность, т.е. долю всосавшегося в организм ЛВ, введенного различными путями, по отношению к его количеству после внутривенного введения. Значительно чаще определяют относительную биодоступность, которая отражает сравнительную оценку всасывания одного и того же ЛВ из испытуемой и стандартной ЛФ. Определение ведут по содержанию ЛВ в крови или в моче после однократного или многоразового введения.
Терапевтическая неэквивалентность ЛВ (ЛФ), изготовленных по различным технологиям (или различными фирмами), зависит от различной их биодоступности. В связи с этим существует понятие биоэквивалентности лекарственных веществ. Биоэквивалентность устанавливают по таким трем параметрам, как максимум концентрации ЛВ в крови, время достижения максимальной концентрации и площадь под кривой изменения концентрации ЛВ в плазме или сыворотке крови, измеренная во времени. Биоэквивалентными называют такие ЛВ, которые обеспечивают одинаковую концентрацию в крови и тканях организма. Нередки случаи, когда аналогичные ЛВ биологически неэквивалентны, так как имеют различную биодоступность. Поэтому при оценке биоэквивалентности следует учитывать важнейшие параметры биодоступности ЛВ. Иными словами, оптимизация лекарственной формы должна осуществляться с точки зрения обеспечения максимально возможной для данного ЛВ степени биодоступности.
Биологическую доступность ЛС можно установить тремя различными путями: методами in vitro с помощью приборов; методами in vivo на животных или у здоровых людей-добровольцев. Установление биологической доступности методами in vitro основано на корреляционной зависимости между скоростью всасывания и скоростью растворения ЛВ. Поэтому для растворимых веществ метод определения скорости растворения служит основным методом определения эффективности высвобождения ЛВ из ЛФ.
Принцип действия созданных для этого многочисленных приборов заключается в механическом разрушении ЛФ и диффузии ЛВ в воду или другую растворяющую среду, имитирующую биологическую жидкость. По мере высвобождения или после полного высвобождения ЛВ растворяющую жидкость удаляют из прибора. Полученные пробы подвергают анализу, используя химические или физико-химические методы. Аналитический контроль — важнейший этап испытания. Лекарственная форма признается соответствующей требованиям скорости высвобождения, если в течение установленного интервала времени из нее переходит в растворяющую жидкость оптимальное количество ЛВ. Следует отметить, что изучение кинетики высвобождения лекарственного вещества in vitro в модельных условиях не может заменить исследования in vivo. Вызвано это различием в механизмах протекающих процессов. Так, при всасывании in vivo вслед за стадией растворения ЛВ следует стадия проникновения через стенки желудка и кишечника. В то же время в условиях in vitro моделируется лишь стадия растворения.
Биологическая доступность методами in vivo устанавливается на лабораторных животных (кроликах, собаках и др.). При этом либо определяют содержание ЛВ (метаболитов) в крови, либо устанавливают скорость их выведения с мочой через определенные промежутки времени. Важнейший этап этих испытаний — количественный анализ. Он усложняется по сравнению с методами in vitro, поскольку приходится анализировать сложную смесь, включающую не только ЛВ или их метаболиты, но и различные соединения, входящие в состав биологических жидкостей.
Для характеристики биодоступности широко применяют способ, основанный на оценке максимальной концентрации ЛВ в крови после введения внутрь изучаемой ЛФ. Такой способ является весьма приблизительным, так как биодоступность зависит не только от степени и скорости всасывания, но и от распределения и элиминации ЛВ в организме.
Для определения биологической доступности у здоровых людей подбирают группы добровольцев определенного возраста и соответствующим образом их готовят: стандартизируются диета, количество выпитой воды, физическая активность, исключается прием других лекарств, возможность стрессовых состояний и т.д. Сущность испытаний заключается в установлении скорости выведения ЛВ с мочой через определенные промежутки времени после введения ЛС. Концентрацию ЛВ или их метаболитов устанавливают с помощью методик биофармацевтического анализа.
Таким образом, одним из основных этапов любого исследования биологической доступности ЛС является использование биофармацевтического анализа для определения концентрации ЛВ (метаболита) в биологических жидкостях.
5. Особенности биофармацевтического анализа
Биофармацевтический анализ — новое перспективное направление фармацевтической химии. Задачей биофармацевтического анализа является разработка способов выделения, очистки, идентификации и количественного определения ЛВ и их метаболитов в таких биологических жидкостях, как моча, слюна, кровь, плазма или сыворотка крови и др. Только на основе применения таких методик можно выполнять биофармацевтические исследования, т.е. изучать вопросы всасывания, транспорта и выведения ЛВ, его биологическую доступность, процессы метаболизма. Все это позволяет предупреждать возможное токсическое воздействие ЛС, разрабатывать оптимальные режимы фармакотерапии и контролировать процесс лечения. Особенно важно определять в биологических жидкостях концентрацию ЛВ, когда они наряду с терапевтическим эффектом проявляют токсичность. Необходимо также контролировать содержание ЛВ в биологических жидкостях больных, страдающих желудочно-кишечными заболеваниями и заболеваниями печени и почек. При таких заболеваниях изменяются процессы всасывания, нарушаются метаболические процессы, замедляется выведение ЛВ из организма.
Биологические жидкости — очень сложные объекты для выполнения анализа. Они представляют собой многокомпонентные смеси, включающие большое число неорганических и органических соединений различной химической структуры: микроэлементы, аминокислоты, полипептиды, белки, ферменты и др. Их концентрация колеблется от 10 мг/мл до нескольких нанограммов. Даже в такой относительно простой физиологической жидкости, как моча, идентифицировано несколько сотен органических соединений. Всякий биологический объект — очень динамичная система. Ее состояние и химический состав зависят от индивидуальных особенностей организма, воздействия факторов внешней среды (состав пищи, физическая и психическая нагрузка и т.д.). Все это еще в большей степени усложняет выполнение биофармацевтического анализа, так как на фоне столь большого количества сложных по химическому строению органических веществ нужно определять нередко очень малые концентрации ЛВ. Вводимые в биологические жидкости ЛВ в процессе биологической трансформации образуют метаболиты, количество которых нередко исчисляется несколькими десятками. Выделение этих веществ из сложных смесей, разделение на индивидуальные компоненты и установление химического состава — задача необычайно трудная.
Следовательно, биофармацевтический анализ представляет собой своеобразный инструмент, необходимый для проведения не только биофармацевтических, но и фармакокинетических исследований.
Вопросы по теме для самостоятельного изучения (для зачета, экзамена), темы рефератов.
1. Изменения, происходящие при лекарственных средств поступлении лекарственных средств в организм. Реакции метаболизма — окисления, гидролиза, восстановления.
2. Влияние различных факторов на фармакокинетику и фармакодинамику лекарственных средств.
Источник


