КОНТРОЛЬ КАЧЕСТВА ЛЕКАРСТВЕННЫХ СРЕДСТВ ПРОМЫШЛЕННОГО ПРОИЗВОДСТВА
Производство ЛС — это серийное получение ЛС в соответствии с правилами организации производства и контроля их качества, утвержденными соответствующим федеральным органом.
Производство ЛС осуществляется предприятиями-производителями, имеющими лицензии на их производство. Государственный контроль производства выполняют федеральный и территориальный органы контроля качества ЛС, в права которых входит:
— беспрепятственный доступ на любое предприятие-производитель ЛС и контрольное изъятие производимых образцов;
— снятие копий с документов, необходимых для проведения контроля производства и качества ЛС;
— запрет производства и продажи уже произведенных ЛС в случаях: 1) если ЛС не прошли государственную регистрацию в РФ (исключение — ЛС, предназначенные для проведения клинических исследований); 2) отсутствия лицензии на производство; 3) изготовления с нарушением правил организации производства и контроля качества ЛС (статья 13 Государственного Закона о лекарственных средствах).
Лицензия на производство ЛС выдается федеральным органом исполнительной власти на срок не менее 5 лет. В случае, если предприятием-производителем изменены условия производства, оно обязано получить новую лицензию на производство.
На фармацевтическом предприятии контроль качества ЛС является частью надлежащей производственной практики, гарантирующей правильность процедуры отбора проб, подготовки пробы к анализу (пробоподготовки), определения действующего вещества, принятия решения о приеме испытуемого материала в соответствии с требованиями, установленными в НД (ФСП, спецификации).
Отдел контроля качества (ОКК) фармацевтического предприятия осуществляет различные типы фармацевтического контроля: входной контроль фармацевтических субстанций и вспомогательных веществ, межоперационный контроль в процессе производства и контроль качества готовой продукции (рис. 6.2).
Таким образом, контроль качества на производстве включает контроль исходного сырья, полупродуктов, лекарственных субстанций и готовых лекарственных форм.
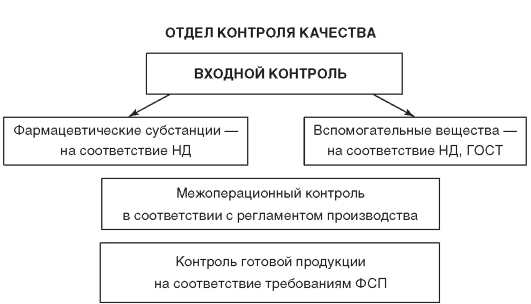
Рис. 6.2. Типы фармацевтического анализа в условиях производства лекарственных средств
(по:Елизарова Т.Е. Современные методы стандартизации и контроля качества лекарственных средств. — М.: МИА, 2008)
В период бурного развития фармацевтической промышленности возникли проблемы качества ЛС, которые не могли быть решены только путем усиления фармакопейного анализа. Обеспечение качества ЛС стало возможным только на базе проведения фармацевтического анализа в соответствии с правилами GMP.
Согласно правилам GMP, объектом контроля становится весь процесс производства ЛС, включая помещения, персонал, документацию. В России правила GMP нашли отражение в виде ГОСТ Р 52249-2009 «Правила производства и контроля качества лекарственных средств».
Стандарт содержит требования к производству и контролю качества ЛС для человека и животных. Стандарт распространяется на все виды ЛС и устанавливает общие требования к их производству и контролю качества, а также специальные требования к производству отдельных видов ЛС. Стандарт не распространяется на обеспечение промышленной безопасности, пожарной безопасности, взрывобезопасности, химической безопасности и безопасности других видов при производстве ЛС, требования к которым приведены в других НД.
Надлежащая практика контроля качества фармацевтических препаратов обеспечивается комплексом мероприятий при их разработке и исследовании с учетом требований GMP. Каждая методика
должна содержать обоснование преимуществ по сравнению с другими в виде представленных результатов сопоставления ее применения (валидация).
Валидация методавключает следующие метрологические характеристики:
— правильность (accuracy) — близость результатов к истинному значению, что может быть проведено при сравнении с результатами, полученными с помощью иной методики, валидированной ранее;
— точность (precision) — согласованность между отдельными результатами испытаний (отклонение отдельных результатов от среднего значения — относительное стандартное отклонение);
— сходимость (repeatability) — точность методики при ее выполнении одним и тем же аналитиком при одних и тех же условиях (реактивы, оборудование, лаборатория);
— воспроизводимость (reproducibility) — точность методики при использовании ее в различных условиях для идентичных образцов, отобранных из одной и той же однородной серии материала (разные лаборатории, исполнители, оборудование, время);
— надежность (robustness) — способность методики давать результаты анализа с приемлемой правильностью и точностью при изменении условий работы для предположительно идентичных образцов из одной и той же однородной серии материала;
— чувствительность (sensitivity) — способность методики испытания регистрировать небольшие изменения концентрации (наклон калибровочной кривой);
— предел обнаружения (limit of detection) — наименьшее содержание, при котором анализируемое вещество может быть обнаружено.
Контроль качества ЛС на отдельных технологических стадиях его получения обеспечивает надлежащее качество конечного продукта.
Для лекарственных препаратов регламентируется надлежащая микробиологическая чистота. Загрязнение микроорганизмами может происходить на разных стадиях производства, поэтому испытания на микробиологическую чистоту проводят на всех стадиях получения ЛС. Основными источниками микробной контаминации являются сырье, вода, оборудование, воздух производственных помещений, упаковка готовой продукции, персонал.
Для количественного определения содержания микроорганизмов в воздухе используют различные методы отбора проб: фильтрацию, осаждение в жидкостях, осаждение на твердые среды. Для оценки микробиологической чистоты выполняют тесты на стерильность.
При определении стерильности ЛС, обладающих выраженным антибактериальным действием, бактериостатическими, фунгистатическими свойствами, а также содержащих консерванты, используют метод мембранной фильтрации.
Метод мембранной фильтрации основан на пропускании ЛС через полимерную мембрану. При этом микроорганизмы остаются на поверхности мембраны. Затем мембрану помещают в соответствующую питательную среду и наблюдают образование колоний при инкубировании.
Также проводят испытания на пирогенность инъекционных препаратов.Необходимость проведения этого теста связана с присутствием в инъекционных препаратах фрагментов клеток грамположительных и грамотрицательных бактерий, грибов, вирусов и эндотоксинов. Применение таких препаратов вызывает жар, озноб, тошноту, иногда летальный исход. Наибольшую опасность представляют эндотоксины, которые являются термостабильными и состоят из липополисахаридов внешней плазматической мембраны грамотрицательных бактерий, попадание которых в организм человека провоцирует резкий воспалительный процесс.
Контроль содержания пирогенных примесей в инъекционных препаратах проводят двумя методами: в опытах in vivo на кроликах и in vitro, с использованием ЛАЛ-реактивов, приготовленных из водного экстракта (лизата) амебоцитов мечехвоста полифема 1 .
ЛАЛ-тест впервые был включен в Фармакопею США в 1980 г., а позже был признан и в европейских странах. В 1997 г. в РФ была утверждена ФС «Определение содержания бактериальных эндотоксинов (ЛАЛ-тест)».
Основными преимуществами ЛАЛ-теста по сравнению с традиционным испытанием на кроликах является возможность оценки уровня бактериальных эндотоксинов в тех препаратах, которые нельзя испытывать на животных; более высокая (в 100 раз) чувствительность ЛАЛ-теста; быстрота выполнения (одно испытание занимает около 1,5 ч); испытания проводит один человек.
Более высокая чувствительность и быстрота ЛАЛ-теста обеспечивают возможность контроля качества воды для приготовления инъекционных ЛС в условиях производства. Этот тест используется для анализа «Воды инъекционной в ампулах», «Раствора натрия хлорида 0,9% для инъекций», различных лекарственных форм инсулина.
Под чувствительностью ЛАЛ-реактива понимают минимальную концентрацию международного.стандарта эндотоксина, которая приводит к образованию плотного геля при реакции с данным ЛАЛ-реактивом в гель-тромб тесте, проведенном при стандартных условиях.
Испытания на кроликах проводят на 12 кроликах породы шиншилла массой 2,5-3,0 кг с соблюдением строгого стандартного рациона питания в специально оборудованном тихом, светлом, без перепадов температуры помещении. Кроликов помещают в определенным образом оборудованные индивидуальные боксы, позволяющие проводить постоянное измерение температуры, которая записывается в автоматическом режиме самописцем, что исключает фальсификацию данных.
В начале опыта препарат вводят 3 животным. ЛС считается апирогенным, если суммарное повышение температуры тела животных (∑Δt) составляет не более 1,2 °С. На втором этапе испытаний препарат вводят 6 животным. ЛС признают пирогенным, если ∑Δt>3,0 °С. На третьем этапе число животных равно 9. ЛС считают пирогенным при ∑Δt>4,5 °С. Наконец, на четвертом этапе используется 12 животных, и препарат считают пирогенным, если ∑Δt>5,4 °С. После окончания проверки составляют протокол, который включает параметры эксперимента, заключение о соответствии проверяемого раствора требованиям на пирогенность.
Метод имеет ряд недостатков. В эксперименте используют животных, чувствительность которых к пирогенам в 3-4 раза ниже, чем у человека. Это требует соответствующего увеличения тест-дозы. Кроме того, многие ЛВ в дозах, близких к терапевтическим, могут вызвать токсические реакции и даже гибель животных,
поэтому используется заниженная величина тест-доз (инфузионные растворы глюкозы, антибиотики — бензилпенициллина натриевая соль, линкомицина гидрохлорид и др.). На проведение одной серии опыта требуется около 5 ч.
1 Мечехвост полифем, краб-подкова, королевский краб. Вид: Limulus polyphemus (Linnaeus, 1758).
LAL(ЛАЛ)-тест (Limulus Amebocyte Lysate) — ферментативная реакция ЛАЛреактива с эндотоксином, в результате которой образуется плотный гель. При отсутствии эндотоксина в анализируемой пробе гель не образуется. Положительный результат ЛАЛ-теста указывает на то, что в исследуемой пробе содержание эндотоксина составляет не менее 0,25 ЕЭ (единиц эндотоксина). Отрицательный результат говорит о том, что в исследуемой пробе содержание эндотоксина — менее
Контрольные вопросы и задания
• Охарактеризуйте этапы фармацевтического анализа: определение подлинности, оценка чистоты (определение примесей), количественный анализ.
• Перечислите правила надлежащей деятельности (GP), в соответствии с которыми должны осуществляться производство и контроль качества ЛС.
• Каковы особенности фармацевтического анализа воспроизведенных ЛС?
• Перечислите типы эквивалентности ЛС. Как производят оценку фармацевтической и биологической эквивалентности?
• Объясните необходимость применения в фармацевтическом анализе стандартных образцов сравнения.
• Перечислите характеристики валидации аналитического метода.
Источник
Тема 10 «Контроль качества мягких лекарственных форм»
учебно-методический материал

Учебный материал для студентов 2 курса
Скачать:
| Вложение | Размер |
|---|---|
| tema_10_kk_mlf.docx | 76.94 КБ |
Предварительный просмотр:
Тема 10: Контроль качества мягких лекарственных форм
1. Общая характеристика мазей и суппозиториев
Мази – мягкая лекарственная форма, предназначенная для нанесения на кожу, раны или слизистые оболочки.
Суппозитории – твердые при комнатной температуре и расплавляющиеся или растворяющиеся при температуре тела дозированные лекарственные формы.
Характер, продолжительность и место приложения действия мазей и суппозиториев зависят от многих биофармацевтических факторов. К наиболее значимым из них следует отнести физико-химические свойства лекарственных и вспомогательных веществ и их концентрация, агрегатное состояние лекарственных веществ, их дисперсность, технология изготовления и др. Это обязывает к строгому подходу в обеспечении качества мазей и суппозиториев и их контролю.
2. Нормативные документы
При изготовлении мазей и суппозиториев в условиях аптеки нормативных документов частного характера нет. Качество лекарственных форм, приготовленных по индивидуальным рецептам, определяется квалификацией провизора, который в своей работе руководствуется действующими нормативными документами общего характера и методическими указаниями
Контроль и заключение о качестве мазей и суппозиториев аптечного изготовления проводится на основании прописи, а также действующих приказов и инструкций.
Нормативным документом, регламентирующим качество мазей или суппозиториев заводского изготовления, является частная фармакопейная статья (ФСП).
3. Формирование показателей качества мазей
Содержание лекарственных веществ в мазях заводского изготовления обычно выражается в относительных единицах (%). В таком случае при использовании в количественном анализе титриметрического метода расчет содержания действующих веществ в мазях осуществляется по формуле:

Q – навеска мази, взятая для анализа, г.
ФС частного характера регламентирует пределы количественного содержания лекарственных веществ, в которые должны укладываться результаты анализа.
Расчет содержания лекарственных веществ в мазях аптечного изготовления осуществляется на общую массу мази по формуле:

Р – общая масса мази по прописи, г
Заключение о количественном содержании лекарственных веществ в мази аптечного изготовления делается на основании расчета относительного отклонения. Оно не должно превышать допустимого отклонения.
На стадии перенесения мази в тару для отпуска (в случае изготовления мази в аптечных условиях), либо при расфасовке мази в склянки или тубы (в случае заводского производства) возможны потери объективного характера, которые будут сказываться на общей массе мази. Вследствие неизбежности этих потерь нормативными документами регламентируется предел 13 отклонений в общей массе мази. Данный показатель определяется нахождением фактической массы мази и последующим расчетом абсолютного и относительного отклонений. Фактическую массу определяют по разнице между результатами взвешивания на технических весах с точностью до 0,01 г мази в таре, предназначенной к отпуску, и тары после удаления из нее мази, промывания в горячей воде и высушивания.
Микробиологическая стабильность мазей обеспечивается соблюдением санитарных норм и правил при их производстве, а также применением консервантов. Однако мази могут быть контаминированы микроорганизмами в процессе транспортировки и хранения. Поэтому ГФ XII регламентирует проведение испытания на микробиологическую чистоту по методике, унифицированной в ОФС «Микробиологическая чистота». Испытание включает количественное определение жизнеспособных бактерий и грибов, а также выявление определенных видов микроорганизмов.
Некоторые мази, например глазные, должны быть стерильными. Их стерильность достигается соблюдением необходимых санитарногигиенических условий и режима стерилизации. Для таких мазей предусматривается испытание на стерильность , о чем должны быть соответствующие указания в ФС. Испытание на стерильность также унифицировано и изложено в ОФС «Стерильность». Наиболее распространенными химическими процессами, приводящими к нарушению стабильности мазей, являются гидролиз и окисление основ. Например, жиры, являясь сложными эфирами высших жирных кислот и глицерина, могут подвергаться гидролизу при наличии в составе мази воды. В результате гидролиза жиров образуются свободные жирные кислоты, которые, в свою очередь, повышают кислотность мази.
Жировые основы могут также подвергаться окислению под воздействием кислорода воздуха на свету или примесей металлов с переменной валентностью.
Образующиеся свободные радикалы ускоряют цепной процесс окисления, что приводит к накоплению различных продуктов с кислородсодержащими функциональными группами.
Продукты деструкции основ, в частности, альдегиды и кетоны придают мази прогорклый запах. Кроме того, они могут оказывать непосредственное раздражающее действие на кожу и слизистые оболочки при нанесении мази. Образующиеся в результате прогоркания основы активные частицы могут инициировать процессы разложения лекарственных веществ в составе мазей.
Таким образом, основными факторами нарушения стабильности мазей являются свет, влага, повышенная температура, содержащиеся в качестве примесей соединения металлов с переменной валентностью и др. Поэтому упаковка и условия хранения мазей должны исключать влияние этих факторов. Мази в соответствии с требованиями нормативных документов отпускают в тубах или банках из стекла, пластмассы и хранят в сухом, прохладном, защищенном от света месте.
Стабильность мази изучают общепринятым методом ускоренного старения. При этом стабильность действующих веществ оценивают по появлению продуктов их разложения, а стабильность основы изучают по процессам гидролиза и окисления с использованием показателей кислотного, перекисного и йодного чисел, определяемых по унифицированным фармакопейным методикам. Проведение данных исследований позволяет определять сроки годности при соблюдении условий хранения и делает возможным исключение этих испытаний при оценке качества готовых мазей. Поэтому в ОФС «Мази» определение кислотного, перекисного и йодного чисел не регламентируется. Однако особое значение при этом приобретает контроль сроков годности, упаковки и условий хранения.
Кроме этого, в блок испытания мазей включают и другие показатели, позволяющие контролировать их стабильность. Так, снижение химической стабильности, в частности, прогоркание жировой основы, приводит к изменению внешнего вида и появлению несвойственного для мази запаха. Нарушение физико – химической стабильности может привести к изменению консистенции мази или ее расслоению. Поэтому, кроме контроля сроков годности и упаковки, необходимо оценивать внешний вид мазей и наличие запаха. Более объективными показателями стабильности действующих веществ будут являться их количественное содержание и отсутствие специфических примесей. В некоторых случаях, кроме вышеназванных испытаний, проводится определение рН водной вытяжки. Это испытание приобретает особую важность при контроле качества глазных мазей ввиду особенностей их применения – нанесения мази на слизистую оболочку глаза.
Таким образом, контроль качества мазей предусматривает:
1. Контроль упаковки, оформления и сроков годности.
2. Контроль внешнего вида мази и запаха.
3. Определение общей массы мази.
4. Определение размера частиц лекарственных веществ в мазях.
5. Определение подлинности лекарственных веществ, входящих в состав мазей.
6. Определение рН водной вытяжки (если в ФС есть указания).
7. Определение специфических примесей.
8. Определение количественного содержания каждого из действующих веществ.
9. Определение микробиологической чистоты мази или стерильности, если в ФС будет на этот счет указание.
4. Формирование показателей качества суппозиториев
Фармацевтическими факторами, в наибольшей мере влияющими на биологическую доступность суппозиториев, являются характер вспомогательных веществ и способ введения лекарственных веществ в суппозиторную основу. Изучение влияния фармацевтических факторов на биологическую доступность является обязательным при обосновании состава суппозиториев и их технологии.
Подготовка лекарственных веществ зависит от их физико–химических свойств, главным образом, растворимости в воде и основе и от количественного содержания в суппозиториях. Для обеспечения высокой дисперсности и лучшего распределения лекарственных веществ в основе предусматривается их измельчение и (или) растворение в подходящем растворителе или части основы. На этих стадиях возможны потери действующего вещества. Для контроля правильности ведения этих операций в блоке испытания суппозиториев предусматривается определение количественного содержания лекарственных веществ. Причем, вследствие неизбежности объективной ошибки нормативные документы регламентируют предел допустимых отклонений в содержании лекарственных веществ.
Подготовленная суппозиторная масса должна быть упруго–пластичной и однородной, без видимых блесток и вкраплений веществ и основы. Однородность суппозиториев определяют визуально на продольном срезе по отсутствию вкраплений. На срезе допускается наличие воздушного стержня или воронкообразного углубления.
Особенностью изготовления суппозиториев является то обстоятельство, что для придания суппозиторной массе требуемых упруго – пластичных свойств к ней добавляются дополнительные вещества. Необходимое количество вязких веществ заранее и точно рассчитано быть не может, а пластичность суппозиторной массы определяется непосредственно в момент ее приготовления. Поэтому масса одного суппозитория в прописи не указывается, а нормативные документы регламентируют предел массы суппозиториев и определение такого показателя как «средняя масса». В соответствии с ОФС данный показатель определяется взвешиванием 20 суппозиториев с точностью до 0,01 г и делением найденной массы на количество суппозиториев в выборке. Предел массы суппозиториев зависит от путей введения. ОФС «Суппозитории» регламентирует для ректальных суппозиториев массу от 0,1 до 4,0 г, для вагинальных от 1,5 до 6,0 г. Широкие пределы колебания массы суппозиториев связаны с возрастом больного, свойствами и количественным содержанием действующих и вспомогательных веществ.
Суппозитории являются дозированной лекарственной формой. Разделение суппозиторной массы на дозы производится не по массе, а по объему. Показатель «средняя масса» не позволяет осуществить контроль стадии дозирования. При этом масса отдельного суппозитория может значительно отличаться от средней массы. Поэтому для контроля стадии дозирования используется дополнительный показатель – отклонение массы отдельного суппозитория от средней массы. Показатель определяется взвешиванием каждого суппозитория в выборке с последующим расчетом абсолютного и относительного отклонений. Абсолютное отклонение рассчитывается с точностью до 0,01 г путем вычитания массы одного суппозитория от средней массы. Затем рассчитывается относительное отклонение, при этом за 100 % принимается средняя масса суппозитория. Данные могут быть оформлены в виде таблицы:

Предел допустимого отклонения в массе суппозиториев регламентируется в ОФС «Суппозитории», в соответствии с которой отклонение не должно превышать ± 5 % и только два суппозитория в выборке могут иметь отклонение ± 7,5 %.
Объективным показателем, позволяющим оценить взвешивание лекарственных веществ и основы, введение лекарственных веществ в основу, их равномерное распределение в ней, разделение суппозиторной массы на дозы является определение количественного содержания лекарственных веществ в одной дозе. Определение всех компонентов в одном суппозитории не всегда возможно из–за их малого содержания. Поэтому для анализа может использоваться несколько суппозиториев, однако расчет количественного содержания каждого из действующих веществ осуществляется на одну дозу.
Таким образом, особенностью количественного определения суппозиториев является то обстоятельство, что на анализ берется целочисленное количество доз. Если расчетная навеска будет составлять нецелочисленное количество суппозиториев, ее приводят к целочисленному значению, используя затем в анализе прием разведения. В любом случае расчет содержания действующего вещества ведется на один суппозиторий:

n – количество суппозиториев, взятых для анализа.
При использовании приема разведения расчетная формула принимает следующий вид:

Для суппозиториев с низким содержанием лекарственных веществ может предусматриваться контроль однородности дозирования , о чем должны быть указания в ФС частного характера.
Для удобства введения суппозиториев в полости тела им придается определенная форма. Ректальные суппозитории могут иметь форму конуса, цилиндра с заостренным концом или иную форму с максимальным диаметром 1,5 см. Вагинальные суппозитории могут быть сферическими (шарики), яйцевидными (овули) или в виде плоского тела с заостренным концом (пессарии). Палочки имеют форму цилиндра с заостренным концом с диаметром не более 1 см. Поэтому специфическими показателями качества суппозиториев являются их размер и форма, которые должны соответствовать пути введения.
Придаваемая суппозиториям форма должна сохраняться при комнатной температуре и не подвергаться деформации в момент введения в полости тела. В то же время при температуре тела суппозитории должны расплавляться или растворяться для высвобождения действующего вещества. Поэтому ход анализа суппозиториев предусматривает проведение испытаний, характеризующих эти свойства. ОФС 22 «Суппозитории» регламентирует несколько унифицированных испытаний, выбор которых определяется характером основы. Для суппозиториев, изготовленных на липофильных основах, определяют температуру плавления и \ или время полной деформации. Определение температуры плавления суппозиториев проводится методом 2а по унифицированной методике, изложенной в ОФС «Определение температуры плавления». Температура плавления суппозиториев не должна превышать нормальной температуры тела. Специфическим показателем качества для суппозиториев является «Определение времени полной деформации». Испытание заключается в определении времени деформации суппозитория под давлением металлического стержня массой 7,5 г при температуре C O 37 ± 1 . Если в ФС частного характера нет других указаний, время полной деформации должно составлять 15 мин.
Для суппозиториев, изготовленных на гидрофильной основе, определяют время растворения по методике, унифицированной в ОФС «Суппозитории». Испытание сводится к определению времени растворения суппозитория при помещении его в сосуд вместимостью 100 мл, содержащий 50 мл воды с температурой C O 37 ± 1 , и периодическом перемешивании через каждые 5 мин. Время растворения должно составлять не более 1 часа.
Стабильность суппозиториев обеспечивается соблюдением санитарных норм и правил при их производстве, а также научно обоснованной упаковкой и условиями хранения. Основными факторами нарушения стабильности суппозиториев являются свет, влага, повышенная температура, примеси металлов с переменной валентностью и др. Поэтому упаковка и условия хранения суппозиториев должны исключать влияние этих факторов. Свечи аптечного изготовления в соответствии с требованиями нормативных документов упаковывают в гофрированные капсулы или колпачки из вощеной или парафинированной бумаги и укладывают в картонные коробки. Суппозитории промышленного производства помещают в контурную упаковку из поливинилхлоридной пленки и укладывают в пачки из картона.
Суппозитории должны храниться в сухом и прохладном месте. Срок хранения суппозиториев, изготовленных в аптеке не должен превышать 10 суток. Срок годности суппозиториев заводского изготовления регламентирован ФС частного характера.
При оценке стабильности суппозиториев обязателен контроль сроков годности, а также целостности упаковки и ее соответствия нормативным документам. Кроме этого, в блок испытания суппозиториев включают дополнительные показатели стабильности: внешний вид суппозиториев и запах. Наиболее объективным показателем стабильности лекарственных веществ в суппозиториях является их количественное содержание.
Суппозитории аптечного изготовления долго не хранятся, в то время как срок годности заводских суппозиториев обычно составляет 2 года. Поэтому в свечах заводского изготовления, содержащих нестабильные лекарственные вещества, ФС частного характера могут рекомендовать определение специфических примесей. Так, например, в суппозиториях 24 ректальных с анальгином для детей ФС регламентирует определение специфической примеси 4–аминоантипирина, которая является токсичной.
Микробиологическая стабильность суппозиториев обеспечивается соблюдением санитарных норм и правил при их производстве, а также применением консервантов. Однако суппозитории могут быть контаминированы микроорганизмами в процессе транспортировки и хранения. ГФ XII регламентирует проведение испытания на микробиологическую чистоту по методике, унифицированной в ОФС «Микробиологическая чистота». Испытание включает количественное определение жизнеспособных бактерий и грибов, а также выявление определенных видов микроорганизмов.
Таким образом, контроль качества суппозиториев предусматривает:
1. Контроль упаковки, маркировки и сроков годности.
2. Контроль формы и ее соответствия пути введения.
3. Контроль средней массы и отклонений массы отдельных суппозиториев от средней массы.
4. Определение однородности.
5. Определение температуры плавления суппозиториев или времени полной деформации.
6. Определение времени растворения.
7. Определение подлинности действующих веществ, входящих в состав суппозиториев.
8. Определение специфических примесей.
9. Определение количественного содержания лекарственных веществ.
10. Определение однородности дозирования, если в ФС есть соответствующие указания.
11. Определение микробиологической чистоты суппозиториев.
Источник


