- Приложение № 5 Тест сравнительной кинетики растворения и сопоставимость профилей растворения
- Тест сравнительной кинетики растворения и сопоставимость профилей растворения
- Общие аспекты теста сравнительной кинетики растворения во взаимосвязи с биоэквивалентностью
- Сопоставимость профилей растворения
- Кинетика растворения лекарственного средства
- Материал и методы
- Результаты исследования
- Обсуждение результатов
- Литература
Приложение № 5 Тест сравнительной кинетики растворения и сопоставимость профилей растворения
Содержимое (Table of Contents)
Тест сравнительной кинетики растворения и сопоставимость профилей растворения
Общие аспекты теста сравнительной кинетики растворения во взаимосвязи с биоэквивалентностью
- При разработке состава лекарственного препарата тест сравнительной кинетики растворения (ТСКР) служит инструментом установления биофармацевтических свойств лекарственного препарата, то есть свойств, способных повлиять на биодоступность. По завершении разработки состава лекарственного препарата и производственного процесса ТСКР используется для контроля качества масштабирования и промышленных серий, чтобы обеспечить как постоянство качества серий, так и сопоставимость профилей растворения с сериями, использованными в опорных клинических исследованиях. Кроме того, в отдельных случаях ТСКР может служить заменой исследованиям биоэквивалентности.
- ТСКР может преследовать различные цели:
а) при экспертизе качества лекарственного препарата:
- для получения характеристик серии, использованной в исследованиях биодоступности (биоэквивалентности) и опорных
- клинических исследованиях, чтобы обосновать спецификации (нормативный документ по контролю качества);
- как инструмент контроля качества серий лекарственных средств в целях подтверждения постоянства производства;
- для получения характеристик референтного лекарственного препарата, использованного в исследованиях биодоступности (биоэквивалентности) и опорных клинических исследованиях;
б) как замена исследованиий биоэквивалентности: чтобы подтвердить (в определенных случаях) аналогичность различных составов исследуемого лекарственного препарата и референтного лекарственного препарата (биовейверы, например, при внесении изменений, изменении состава в ходе разработки лекарственного препарата и воспроизведенные лекарственные препараты, в соответствии с требованиями раздела IV Правил проведения исследований биоэквивалентности лекарственных препаратов в рамках Евразийского экономического союза, утверждаемых Евразийской экономической комиссией и приложения № 4 к Правилам проведения исследований биоэквивалентности лекарственных препаратов в рамках Евразийского экономического союза, утверждаемых Евразийской экономической комиссией);
чтобы установить постоянство качества серий лекарственных препаратов (исследуемого и референтного лекарственного препарата), на которых будет основываться выбор соответствующих серий для использования в исследованиях in vivo.
- Методы испытаний необходимо разработать применительно к каждому лекарственному препарату на основании общих и (или) частных фармакопейных требований. Если указанные требования не удовлетворительны и (или) не отражают процесс растворения и всасывания in vivo (биорелевантность), допустимо использовать альтернативные методы, при условии наличия у них достаточной дискриминационной способности, то есть способности улавливать разницу между сериями с приемлемой и неприемлемой биодоступностью лекарственного препарата в условиях in vivo. Необходимо всегда принимать во внимание современные сведения (включая взаимодействие характеристик лекарственного препарата, основанных на биофармацевтической системе классификации и вид лекарственной формы.
- Для того чтобы получить полноценные профили растворения, интервалы между отбором проб должны быть достаточно частыми (не реже чем через каждые 15 минут). В период максимального изменения профиля растворения отборы проб рекомендуется осуществлять еще чаще. Для построения правильного профиля растворения быстро растворяющихся лекарственных препаратов, полное растворение которых укладывается в 30 минут, отборы проб необходимо осуществлять каждые 5 или 10 минут.
- Если действующее вещество является хорошо растворимым, допускается предположение, что проблемы с биодоступностью не возникнут, если в дополнение к этому лекарственная форма быстро растворяется при физиологических значениях pH, а вспомогательные вещества не влияют на биодоступность. Напротив, если действующее вещество ограниченно растворимо или малорастворимо, фактором, лимитирующим скорость всасывания, может стать растворимость лекарственной формы. Аналогичная ситуация возникает, если вспомогательные вещества влияют на высвобождение и последующее растворение действующего вещества. В таких случаях необходимо проводить ТСКР в различных условиях с соответствующей схемой отбора проб.
Сопоставимость профилей растворения
6. Результаты ТСКР и основанные на них выводы (например, в обоснование биовейвера) признаются правильными, если построение профиля растворения основывалось на достаточном количестве временных точек.
7. В дополнение к требованиям, изложенным в разделе I настоящего приложения, в отношении лекарственных форм с немедленным высвобождением необходимо провести сравнение во временной точке «15 минут», чтобы выяснить, произошло ли полное растворение до опорожнения желудка.
Если в течение 15 минут растворилось более 85 % действующего вещества (от номинального количества), профили растворения признаются сопоставимыми без дальнейшей математической обработки данных.
Если 85 % действующего вещества растворилось в течение 30, а не 15 минут, то необходимы 3 временные точки: до истечения 15 минут, на 15-й минуте и в точке, когда степень высвобождения составляет около 85 %.
- Рекомендации по лекарственным препаратам с модифицированным высвобождением изложены в соответствующих документах Союза.
- Сопоставимость профилей растворения может быть определена с использованием фактораf по следующей формуле:
f — фактор подобия (сходимости);
n — количество временных точек;
Qfi(t) — среднее значение степени высвобождения (в процентах) действующего вещества в точке t [после начала исследования] из референтного лекарственного препарата;
Qr(t) — среднее значение степени высвобождения (в процентах) действующего вещества в точке t [после начала исследования] из исследуемого лекарственного препарата.
При использовании данной формулы необходимо определить степень высвобождения действующего вещества из исследуемого лекарственного препарата и референтного лекарственного препарата.
- Оценка фактора подобия (сходимости) основана на следующих условиях:
- а) минимальное количество временных точек — 3 (не считая нулевой точки отбора);
- б) для обоих сравниваемых лекарственных препаратов выбираются одинаковые временные точки;
- в) для каждой временной точки необходимо минимум 12 значений степени высвобождения действующего вещества для обоих лекарственных препаратов;
- г) для каждого из составов допускается не более одного случая превышения среднего значения степени высвобождения 85 %;
- д) относительное стандартное отклонение (коэффициент вариации) для степени высвобождения действующего вещества в первой временной точке любого из лекарственных препаратов не должно превышать 20 %, а во всех последующих — не более 10 %.
- Критерий приемлемости для фактора подобия f2) составляет от 50 до 100, что подтверждает сопоставимость профилей растворения.
В случае несоответствия критерию приемлемости по f профили растворения можно сравнивать, используя альтернативные методы (например, расчет фактора различия f>, функцию распределения Вейбулла или сравнение степеней высвобождения в разных временных точках (например, по t-критерию Стьюдента)).
- Методы, альтернативные расчету по f2, считаются приемлемыми, если они статистически корректны, а их использование достаточно обосновано.
- Необходимо заранее определить и обосновать пределы приемлемости критерия сопоставимости, но при этом они не должны превышать 10 %. Кроме того, вариабельность растворения между данными исследуемого и референтного лекарственного препарата также должна быть сопоставимой, однако более низкая вариабельность для исследуемого лекарственного препарата является приемлемой.
Необходимо представить обоснование, что статистическое программное обеспечение прошло валидацию.
Необходимо дать подробное описание и объяснение всем действиям, предпринятым в ходе исследования, с представлением соответствующих обобщающих таблиц.
Источник
Кинетика растворения лекарственного средства
Кафедра клинической фармакологии ГБОУ ВПО РНИМУ им. Н.И. Пирогова Минздрава России, Москва
Цефиксим является полусинтетическим цефалоспорином III поколения для перорального применения. Препарат был разработан в конце 1980-х гг. японской фармацевтической компанией Fujisawa, которая после слияния с компанией Yamanouchi была преобразована в Астеллас Фарма.
Цефиксим обладает высокой активностью в отношении грамотрицательных бактерий, таких как Haemophilus influenzae, Moraxella catarrhalis, представителей семейства Enterobacteriaceae (включая Escherichia coli, Klebsiella spp. и Proteus spp.), Neisseria gonorrhoeae [1]. Следует отметить, что в отношении грамотрицательной флоры активность цефиксима несколько выше, чем в отношении грамположительных микроорганизмов. Тем не менее препарат проявляет высокую активность в отношении β-гемолитических стрептококков группы А и В и пневмококков, чувствительных к пенициллину [2]. Цефиксим применяется для лечения широкого круга заболеваний, включающих инфекции верхних и нижних дыхательных путей, инфекции мочевых путей; неосложненную гонорею.
Следует отметить, что на фармацевтическом рынке России доля генериков является очень высокой и составляет от 78 до 95% [3]. При этом данные ряда исследований, проведенных в последнее время в России и за рубежом, ставят под сомнение качество воспроизведенных препаратов (генериков), зарегистрированных в Российской Федерации [4, 5].
Представляют интерес результаты исследования П.А. Ламберт и соавт. (2004), в котором изучалась фармацевтическая эквивалентность оригинального (референтного) цефтриаксона (Роцефин®) и 34 генериков [4]. Были выявлены существенные различия между оригинальным препаратом и генериками. Так, в 18 случаях были выявлены нарушения Европейской и Американской фармакопеи. Наиболее частыми отклонениями были нарушение прозрачности раствора и наличие примесей тиотриазина (продукт разрушения цефтриаксона), при этом 4 генерических препарата не были стерильными [4].
Значимые различия в эффективности оригинальных антибиотиков и генериков при лечении тяжелой внебольничной пневмонии были выявлены в ретроспективном исследовании Н.Ю. Векслера (2012) [6]. Пациенты первой группы получали оригинальные препараты (комбинацию оригинального препарата цефтриаксона с кларитромицином) или левофлоксацин в рекомендованных дозах. Больные второй группы – генерик цефтриаксона плюс кларитромицин либо воспроизведенный левофлоксацин в тех же дозировках. Оказалось, что у пациентов, получавших оригинальные препараты, длительность пребывания в отделении реанимации и интенсивной терапии (ОРИТ) составляла в среднем 6,4±0,2 дня, при этом пациенты, получавшие воспроизведенные препараты, находились в ОРИТ значительно дольше – 20,16±0,8 суток (p≤0,01). Следует также отметить, что среди пациентов, получавших генерики, летальность была выше и составляла 27,5%, у тех же, кто получал оригинальные препараты, величина данного показателя составляла 18,5%. Таким образом, несмотря на ограничения данного исследования, оно продемонстрировало недостаточную эффективность генериков по сравнению с оригинальными препаратами у госпитализированных пациентов с тяжелой внебольничной пневмонией. Наряду с этим, по мнению автора работы, массовое использование воспроизведенных препаратов будет способствовать селекции антибиотикорезистентности возбудителей инфекций [6].
В связи с вышеперечисленными данными особую актуальность приобретают исследования, целью которых является изучение различных генериков лекарственных средств, присутствующих на фармацевтическом рынке Российской Федерации.
Одним из методов оценки качества твердых лекарственных форм препаратов является сравнительный тест кинетики растворения, который позволяет оценивать скорость и степень высвобождения in vitro активного компонента и является одним из важнейших критериев оценки качества. Фактически его использование при анализе лекарственного препарата и есть испытание, которое наряду с оценкой фармацевтической эквивалентности позволяло бы проводить предварительную оценку биоэквивалентности воспроизведенного лекарственного средства [7].

Целью работы было проведение теста сравнительной кинетики растворения двух препаратов – цефиксима Супракс Солютаб® (таблетки диспергируемые 400 мг, «Астеллас Фарма Юроп Б.В.», Нидерланды) и препарата Панцеф® (таблетки, покрытые пленочной оболочкой, 400 мг, «АЛКАЛОИД АО» Республика Македония).
Материал и методы
Все исследования были выполнены в испытательной лаборатории ООО «Национальное агентство клинической фармакологии и фармации». Исследование кинетики растворения вышеперечисленных лекарственных препаратов цефиксима проводили при различных значениях рН среды: 1,2 (имитирует кислотность в желудке натощак); 4,5 (имитирует значение pH в верхнем отделе тонкой кишки); 6,8 (имитирует pH в среднем отделе тонкой кишки) и 7,2 (требования нормативной документации). Растворение таблеток осуществлено в соответствии с основной методикой и требованиями Государственной фармакопеи XI (выпуск 2, с. 158) на лопастной мешалке со скоростью вращения 100 оборотов в минуту при температуре 37±0,5°С, объем среды растворения составил 900 мл. Определение проводили в соответствии с Общей фармакопейной статьей 42-0003-00 на тестере растворения фирмы «Varian» модели VK 7025.
Образцы объемом по 10 мл отбирали через заданные промежутки времени: 5, 10, 15, 30, 45 и 60 минут. Определение количества растворившегося цефиксима проводили спектрофотометрическим методом на УВИ-спектрофотометре модели Cary 100 фирмы «Varian».
Для количественной оценки эквивалентности кинетики растворения лекарственных препаратов Супракс Солютаб® и Панцеф® использовали Руководство по экспертизе лекарственных средств (том III, 2014) [8]. В соответствии с этими указаниями если высвобождение вещества происходит в течение 15 минут, то вещества эквивалентны. Количественно эквивалентность кинетики растворения лекарственного средства оценивают из фактора сходимости v (для веществ со временем высвобождения более 15 минут). Кинетика растворения считается эквивалентной, если значение параметра f2 лежит в пределах от 50 до 100.
Результаты исследования
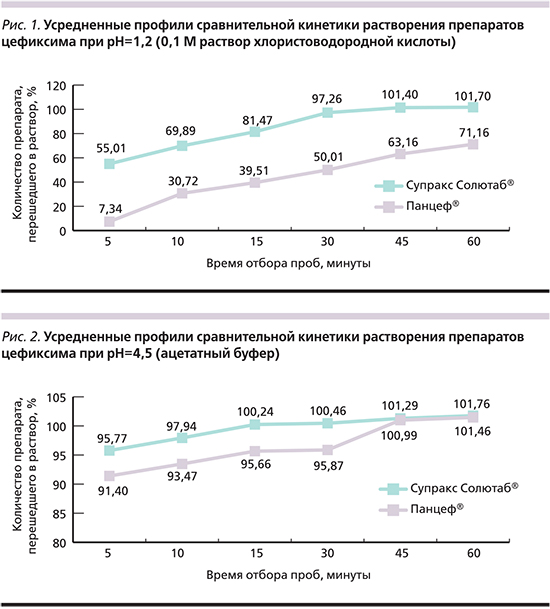
На основании полученных данных были построены графики кинетики растворения препаратов Супракс Солютаб® и Панцеф® (рис. 1–4).
Следует отметить значительную вариабельность кинетики растворения препарата Панцеф®, зависящую от кислотности среды высвобождения. Так, при рН среды, равном 1,2, уже в течение первых 15 минут более 80% цефиксима из препарата Супракс Солютаб® переходит в раствор, в то же время количество действующего вещества, перешедшего в раствор из препарата Панцеф®, оказалось в 2 раза меньше и не превышало 40% (рис. 1). Через 1 час 101,7% активного вещества из препарата Супракс Солютаб® перешло в раствор, при этом величина данного показателя у генерика составила 71,16%. Таким образом, более 28% действующего вещества препарата Панцеф® не перешло в раствор, что в условиях in vivo может сопровождаться снижением биодоступности препарата и приводить к созданию субтерапевтических концентраций в очаге инфекции.

Было установлено, что при рН=4,5 кинетика растворения обоих изученных препаратов была сопоставимой. Так, в течение первых 15 минут теста более 95% цефиксима из препарата Панцеф® перешло в раствор, при этом доля препарата, перешедшего в раствор из препарата Супракс Солютаб®, составила 100,46%. Следует отметить, что через 1 час более 100% цефиксима из обоих изучаемых препаратов перешли в раствор (рис. 2).
Проведение теста растворения при значениях рН=6,8 и 7,2 продемонстрировало значительные различия между препаратами Супракс Солютаб® и Панцеф®. Через 1 час после начала теста при заданных значениях рН количество цефиксима, перешедшего в раствор, у препарата Супракс Солютаб® превышало аналогичные показатели у Панцеф® на 20% (рис. 3–4).
На основании полученных результатов также был рассчитан фактор сходимости (см. таблицу).
Из значений фактора подобия, приведенных в таблице, следует, что кинетика растворения исследуемого препарата Супракс Солютаб® (таблетки диспергируемые 400 мг, «Астеллас Фарма Юроп Б.В.», Нидерланды) в трех средах (фосфатного буфера при pH=7,2 – среда НД, средах 0,1 М хлористоводородной кислоты – рН=1,2, фосфатного буфера – pH=6,8) не эквивалентна кинетике растворения препарата сравнения Панцеф® – таблетки, покрытые пленочной оболочкой, 400 мг («АЛКАЛОИД АО», Республика Македония). При этом следует обратить внимание на то, что количество растворенного цефиксима через 1 час при исследовании кинетики растворения препарата Панцеф® было на 20–30% меньше, чем при изучении препарата Супракс Солютаб®. Несоответствие кинетики растворения может быть «прогностическим признаком» недостаточной биодоступности генерика и как следствие – недостаточной клинической эффективности препарата.
Обсуждение результатов
Результаты нашего исследования показали значительные различия кинетики растворения между протестированными препаратами диспергируемыми таблетками Супракс Солютаб® и препаратом сравнения – таблетками, покрытыми оболочкой, Панцеф®. Было установлено, что препараты Супракс Солютаб® и Панцеф® не эквивалентны по кинетике растворения в трех средах. Наряду с этим высвобождение активного компонента цефиксима в препарате Супракс Солютаб® до 80% за 15 минут происходит во всех исследуемых средах (среда НД, среда 0,1 М хлористоводородной кислоты рН=1,2, фосфатного буфера pH=6,8, ацетатный буфер рН=4,5), в то же время высвобождение 80% действующего вещества за 15 минут у препарата Панцеф® происходит лишь при рН среды, равном 4,5. Таким образом, было установлено, что препарат Супракс Солютаб® обладает более стабильной по сравнению с генериком Панцеф® кинетикой растворения с меньшей зависимостью от кислотности среды.
Выявленные in vitro различия между тестируемыми препаратами имеют большое значение в клинике. В настоящее время доказано, что высокую эффективность антимикробной терапии при использовании β-лактамов можно ожидать в ситуации, когда концентрация антибиотика в очаге инфекции превышает величину его минимальной подавляющей концентрации (МПК) в отношении возбудителя в течение 40–50% времени от интервала между введениями препарата (T>МПК) [9]. Учитывая выявленную зависимость кинетики растворения препарата Панцеф® от рН среды можно предположить, что in vivo возможна ситуация, в которой до 30% активного вещества не перей-дет в раствор и, соответственно, не поступит в системный кровоток. Все это будет сопровождаться уменьшением концентрации антибиотика в очаге инфекции, что в конечном итоге может приводить к неэффективности антимикробной терапии и селекции резистентных штаммов микроорганизмов [10].
Таким образом, полученные нами данные свидетельствуют о том, что кинетика растворения препарата Панцеф® не эквивалентна таковой препарата Супракс Солютаб® при значениях рН среды 1,2; 6,8 и 7,2. В связи с этим использование генерика в определенных ситуациях может сопровождаться увеличением риска клинических неудач и ростом антибиотикорезистентности.
Литература
1. Brogden R.N., Campoli-Richards D.M. Cefixime. A review of its antibacterial activity. Pharmacokinetic properties and therapeutic potential. Drugs. 1989;38:524–50.
2. Grayson M.L., et al. Kucer s use of antibiotics. 6-th ed., 2010;1:3049.
3. Белоусов Ю.Б. Дженерики – мифы и реалии. Remedium. 2003;7–8:4–9.
4. Ламберт П.А., Конвей Б.Р. Сравнение фармацевтического качества генерических препаратов цефтриаксона и роцефина. КМАХ. 2004;6:260–72.
5. Васильева Н.В., Блинов Н.П., Выборнова И.В. Чувствительность Candida species к флуконазолу и некоторым его дженерикам в испытаниях in vitro. Проблемы медицинской микологии. 2002;4:43–4.
6. Векслер Н.Ю. К вопросу об эффективности антибиотиков-генериков в интенсивной терапии тяжелой внебольничной пневмонии. КМАХ. 2012;14:167–69.
7. Зырянов С.К., Белоусов Ю.Б., Камаев А.В., Лелишенцев А.А., Зверков Ю.Б. Эффективность применения левофлоксацина – слагаемые успеха. КМАХ. 2012;14:34–7.
8. Миронов А.Н. Руководство по экспертизе лекарственных средств. Том III. М., 2014.
9. Auckenthaler R. Pharmacokinetics and pharma-codynamics of oral beta-lactam antibiotics as a two-dimensional approach to their efficacy. J. Antimicrob. Chemother. 2002;50(Suppl.):13–7.
Источник


