- Кинетика лекарственных веществ, ее основные параметры и их фармакотера-певтическое значение. Пути направленной регуляции фармакокинетики лекарственных средств.
- Фармакокинетика
- Содержание
- Фармакокинетика [ править | править код ]
- Однокомпартментная модель [ править | править код ]
- Двухкомпартментная модель [ править | править код ]
- Подход без использования компартментного принципа [ править | править код ]
- Расчет дозировки [ править | править код ]
Кинетика лекарственных веществ, ее основные параметры и их фармакотера-певтическое значение. Пути направленной регуляции фармакокинетики лекарственных средств.
ФАРМАКОКИНЕТИКА греческое слово, PHARMACON — лекарство, KINEO — двигать. ФАРМАКОКИНЕТИКА (ФК) — это один из основных разделов фармакологии, изучающий движение лекарств, а именно она в количественном плане описывает (характеризует) абсорбцию (всасывание), распределение, биотрансформацию и экскрецию (выведение) лекарственных средств из организма. ФК — изучает пути прохождения и изменения лекарственных средств в организме, а также зависимость от этих процессов эффективности и переносимости препаратов. Фармакокинетика позволяет оценить динамику концентрации лекарственных средств в организме. Фармакокинетические исследования позволяют оценить процессы всасывания (абсорбции), распределения, связывания с белками, биотрансформации и выведения из организма лекарственных средств. Полученные в результате этих исследований данные создают ту качественную и количественную основу, с помощью которой можно прогнозировать степень попадания лекарственного вещества к месту его действия.
В свою очередь, эти данные необходимы для научно обоснованного выбора рациональных дозировок, путей и схем применения лекарственных средств для обеспечения наиболее эффективного лечения больных и предупреждения побочных эффектов и передозировок.
15. Механизмы резорбции лекарственных веществ. Факторы, влияющие на полноту и скорость всасывания при энтеральном способе введения. Биологическая доступность как критерий фармакотерапевтической эффективности.
Энтеральный (через желудочно-кишечный тракт) — это сублингвальный, пероральный, через зонд в желудок и 12-престную кишку, ректальный.
1- Пассивная диффузия,протекает по градиенту концентрации /из большей в мень-шую) и без затрат энергии. Она может идти через водные порты (в таком случае она называется фильтрация). Пассивной диффузией через мембраны клеток по градиенту концентрации путем растворения в липидной основе протекают жирорастворимые полярные и неполярные молекулы многих лекарственных веществ и ядов: этанола, эфипа, фторотана.
2. Облегченная диффузия — транспорт лекарственных средств через мембраны с участием молекул специфических переносчиков. Этот вид транспорта также происходит по градиенту концентрации, но скорость его выше. Так транспортируются ионы, метаболиты, некоторые витамины. Типичные примеры облегченной диффузии — это всасывание витамина В12 (цианокобаламина) с помощью гастромукопротеида («внутренний фактор Кастла») или всасывание железа с помощью белка апоферритина.
3. Активный транспорт — это перенос молекул лекарственного,вещества против градиента концентрации и электрического градиента, сопряженный с затратой энергии. Энергетические затраты обеспечиваются за счет гидрблиза молекул АТФ . Для активного транспорта характерна: структурная специфичность, насыщаемость системы транспорта и возможность ее конкурентного торможения. Примеры: всасывание в ЖКТ Na, К, Са с помощью Na-K-АТФ-азы, глюкозы, аминокислот, витаминов группы В, транспорт йода в щитовидную железу.
4..Пиноцитоз — это проникновение лекарственных средств путем, инвагинации поверхности биомембраны с последующим образованием везикулы вокруг транспортируемого вещества и проникновением его в циоплазму. С помощью пино- цитоза в клетку приникают крупные белковые молекулы.
Для оказания терапевтического эффекта лекарственное вещество должно быть доставлено в те органы или ткани, в которых осуществляется его специфическое действие (в биофазу). При внутрисосудистом введении лекарство сразу и полностью попадает в кровеносное русло. При других путях введения (перорально, в/м, п/к и т. д. ) прежде чем попасть в кровоток, лекарственное вещество должно пройти ряд биологических мембран клеток (слизистой желудка, клеток печени, мышц и т. д. ) и только тогда какая-то часть его попадет в системный кровоток. Эффект препарата во многом зависит от того, какая часть от введенной дозы лекарственного средства попадает в системный кровоток. Этот показатель характеризует биологическую доступность средства (F). Таким образом, посуществу, биодоступность лекарства отражает концентрацию его у рецепторов, то есть в крови и тканях организма после всасывания. Естественно, что биодоступность одного и того же средства будет разная у каждого больного. Очевидно, что при внутивенном введении лекарства биодоступность его равна приблизительно 100%, а при других путях введения биодоступность почти никогда не достигает 100%.
Различают АБСОЛЮТНУЮ И ОТНОСИТЕЛЬНУЮ БИОДОСТУПНОСТЬ . Абсолютная биодоступность — это доля поглощенного препарата при внесосудистом введении по отношению к его количеству после в/венного введения.
Важным показателем является ОТНОСИТЕЛЬНАЯ БИОДОСТУПНОСТЬ, которая определяет относительную степень всасывания лекарственного вещества из испытуемого препарата и из препаратов сравнения. Другими словами, относительная биодоступность определяется для различных серий препаратов, для лекарственных средств при изменении технологии производства, для препаратов, выпущенных различными производителями, для различных лекарственных форм. Для определения относительной биодоступности могут использоваться данные об уровне содержания лекарственного вещества в крови или же его экскреции с мочой после одноразового или многократного введения. Этот термин важен при сравнении 2-х препаратов между собой.
Источник
Фармакокинетика
Содержание
Фармакокинетика [ править | править код ]
Предметом фармакокинетики является анализ всех факторов, определяющих абсорбцию, распределение, метаболизм и экскрецию лекарственных веществ. Термин отражает скорость перемещения (кинетика) лекарств (фармако) в организме и вне его.
Однокомпартментная модель [ править | править код ]
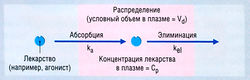
Согласно простейшей модели, организм рассматривается как единое однородное пространство (компартмент), в которое лекарство вводят и из которого оно элиминируется (рис. 4.10). Предполагается, что введенное лекарство сразу же распределяется по всему пространству. Если элиминация является кинетическим процессом первого порядка, то ее скорость пропорциональна концентрации в плазме, т.е. лекарство выводится экспоненциально. Математическое уравнение для этого экспоненциального соотношения выглядит так:
Ct = С0 • e_kelt, (ф. 4.2)
где Ct — концентрация в плазме в данное время (t), С0 — вычисленная начальная концентрация в плазме при t = 0, kel — константа скорости элиминации, е — основание натурального логарифма. Если взять логарифм концентрации в плазме, экспоненциальный процесс выглядит в виде прямой линии (рис. 4.11). Наклон линии на рис. 4.11 в действительности составляет kel/2,303. Деление на 2,303 необходимо потому, что концентрация в плазме дана в десятичном логарифме, а не в логарифме с основанием е. Такое представление использовано на рис. 4.11, т.к. десятичное основание более знакомо фармакологам. Если начальную экстраполированную концентрацию лекарства (С0) в компартменте разделить на введенную дозу, получают теоретический объем, необходимый для описания дозы лекарства. Это пространство называют условным объемом распределения (Vd).
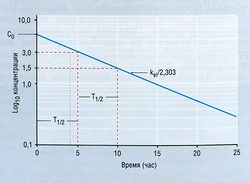
В то время как константа скорости элиминации показывает, как быстро элиминируется лекарство, расчет с использованием kel позволяет определить Т1/2, т.е. время, в течение которого концентрация лекарства в плазме падает на 50%. Поскольку падение концентрации лекарства со временем в логарифмическом исчислении является линейным, величина Т1/2 будет постоянной независимо от концентрации лекарства. Отношение kel и Т1/2 описывает уравнение:
Т1/2 х kel = 0,693 (ф. 4.3)
Поскольку величина T1/2 связана с Vd, значение Т1/2 не всегда отражает способность организма элиминировать лекарство. Предпочитаемый кинетический термин, показывающий способность организма удалять лекарство из кровотока, — это клиренс плазмы (Clp), равный Vd X kel. Клиренс остается постоянным для большинства лекарств, если механизмы элиминации не изменяются под влиянием патологических процессов и/или физиологических факторов.
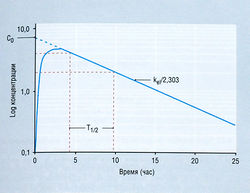
На рис. 4.11 показано снижение концентрации лекарства в плазме после его в/в введения. Однако при пероральном приеме необходимо время для абсорбции, и кривая снижения концентрации напоминает картину, изображенную на рис. 4.12. Форма кривой соотношения концентрации лекарства в плазме и времени отражает взаимодействие двух процессов первого порядка: поступления (абсорбции) вещества в одиночный кинетический компартмент и исчезновения (элиминации) вещества из него. Расчет Vd, Т1/2, kel и Сlр тот же самый, как после в/в введения, однако некоторые из производных параметров (Vd и Сlр) будут возрастать, если происходит значительная потеря лекарства до абсорбции (предсистемная элиминация вещества). Эта ситуация возникает в результате того, что концентрация вещества в плазме уменьшается вследствие предсистемной элиминация вещества, поэтому определение инициальной концентрации лекарства (С0) дает сниженную величину. Эта простая модель единственного однородного компартмента с кинетикой первого порядка вполне пригодна для расчета режима дозировки большинства лекарств.
Двухкомпартментная модель [ править | править код ]
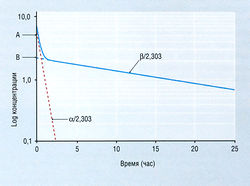
Для некоторых лекарств график, изображающий соотношение логарифма концентрации в плазме и времени, имеет криволинейную форму (рис. 4.13). Чтобы объяснить это явление, требуется расширить однокомпартментную модель. В простейшем виде организм следует рассматривать как двухкомпартментную модель (рис. 4.14). С определенным основанием можно считать, что объяснить криволинейный график на рис. 4.13 можно двумя линейными процессами. Эти два процесса известны под названием а- и β-фаз и характеризуются соответствующими для каждой из них константами скорости элиминации и Т1/2. Согласно этой модели, константа скорости кβ конечной линейной фазы (β) не та, что у kel на рис. 4.11 (модель одного компартмента). β-Фаза представляет собой более медленный процесс. Чем больше расхождение между кβ и kel, тем больше ошибка, если считать правомерной модель одного компартмента. К счастью, для большинства лекарств расхождение между кβ и kel не так велико, как межиндивидуальные различия кинетики, поэтому открытая модель одного компартмента служит приемлемой клинической аппроксимацией для индивидуального выбора дозировки.
Очевидно, что как одно-, так и двухкомпартментные модели дают чрезмерно упрощенное представление о распределении лекарств в организме, где лекарственные вещества абсорбируются и удаляются посредством метаболизма и экскреции. Когда расхождения в модельном распределении лекарств становятся клинически очевидными для данного лекарства, его применение следует рассматривать как особый случай более сложного соотношения распределение-эффект. Лекарства, подвергаемые дозозависимому распределению (например, фенитоин, аспирин), относятся к этой специальной категории более сложного соотношения между дозой, концентрацией и фармакологическими эффектами.

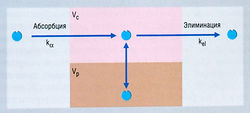
Описание к Рис. 4.13 Изменение концентрации лекарства в плазме в зависимости от времени после одномоментного внутривенного введения: открытая двухкомпартментная фармакокинетическая модель. Константа скорости конечной фазы III отличается от kel, и снижение концентрации со временем отражает более сложное соотношение между распределением и элиминацией лекарства. Начальное более быстрое падение концентрации циркулирующего лекарства (а) отражает в основном его перераспределение в периферический компартмент (Vp) (см. рис. 4.14) плюс незначительный компонент элиминации. Конечная, выраженная прямой линией фаза, отражающая зависимость концентрации в плазме от времени, представляет собой композит элиминации лекарства, замедленной возвратом вещества из Vp в центральный компартмент, в котором вещество распределяется быстро (Vc); тем самым снижается видимая скорость удаления лекарства из плазмы. Эту фазу обозначают как фазу кр. Обратная экстраполяция на время «ноль» дает отрезок, отсекаемый на координатной оси в точке В. Экстраполированное значение кр вычитают из наблюдаемой концентрации лекарства в то же самое время после его введения и откладывают на графике оставшуюся величину. Линия регрессии, проведенная через эти точки, дает наклон к„ и отрезок, отсекаемый на координатной оси в точке А, отражающий распределение лекарства в Vp.
Описание к Рис. 4.14 Схема открытой двухкомпартментной фармакокинетической модели, отражающая концентрации лекарства после внутривенного введения. При одномоментном (болюсном) в/в введении величина к„ несущественна, и в модели ею можно пренебречь. Если лекарство вводят путем инфузии, ка представляет собой константу нулевого порядка, равную скорости инфузии вещества. Существует центральный компартмент, в котором лекарство распределяется быстро (Vc) и из которого оно элиминируется, и периферический компартмент, в котором лекарство может распределяться (Vp), а затем возвращаться, выравнивая изменения концентрации вещества в центральном компартменте, когда лекарство элиминируется. kel — константа элиминации.
Описание к Рис. 4.15 Уравнения фармакокинетического распределения лекарства в отсутствие компартментализации. Константа конечной скорости распределения здесь обозначена А,, и ее интерпретация отличается от таковой для двух моделей, представленных на рис. 4.10-4.14. Введен новый расчет, называемый площадью под кривой (ППК), соответствующей соотношению между концентрацией в плазме и временем. С1р — клиренс плазмы; Т1/2 — период полувыведения; Vd — условный объем распределения.
Продолжительность действия лекарств
- Клинически более важно знать действие лекарства во времени, а не фармакокинетику его концентрации. Между действием лекарства (эффекторная концентрация) и концентрацией в крови может существовать (а может и не быть) линейное или простое соотношение
- Время, необходимое для проявления эффектов лекарства, может быть гораздо длительнее, чем действия лекарства на его молекулярную мишень, т.к. система реагирования имеет собственные временные ограничения
- Например, β-блокаторы очень быстро замедляют частоту сердечных сокращений, тогда как для проявления антикоагулирующего действия варфарина требуется несколько дней
Подход без использования компартментного принципа [ править | править код ]
Этот подход используют для упрощенного определения фармакокинетических параметров, на основании которых можно рассчитать дозу лекарства. Многое в этом подходе заимствовано из открытой однокомпартментной модели. Площадь под кривой (ППК) концентрации представляет в интегральном виде соотношение между концентрацией в плазме и временем, и расчет кинетических параметров проводят соответственно уравнениям, приведенным на рис. 4.15. Этот метод до некоторой степени компенсирует экстраполяцию концентрации лекарства на С0 в модели одного компартмента, когда лекарство еще не абсорбировалось в организме после перорального приема.
Расчет дозировки [ править | править код ]
Большинство лекарств применяют длительно. Для лекарств с элиминацией первого порядка общее количество вещества в организме возрастает, пока экскретируемое количество не становится равным введенной дозе в расчете на единицу времени, т.е. концентрация в плазме достигает стационарного состояния. Время достижения этого состояния для таких лекарств зависит только от конечного Т1/2. Расчет показывает, что 94 и 97% стационарного состояния создаются через 4 и 5 Т1/2 соответственно. Для практических целей принимают, что стационарное состояние в это время существует (рис. 4.16). Чем чаще вводят дозу лекарства, тем выше его количество в организме при стационарном состоянии и тем меньше вариация между концентрацией в плазме на пике и минимальной концентрацией. Чем реже вводят лекарство, тем ниже его количество в организме при стационарном состоянии и тем больше различие между концентрацией в плазме на пике и минимальной концентрацией. Если интервал между введениями больше, чем 2 Т1/2, накопление лекарства при длительном приеме внутрь считается клинически несущественным.
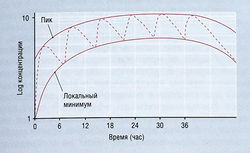
- Большинство лекарств элиминируется из организма как постоянная фракция их концентрации в плазме (процесс первого порядка)
- Время достижения стационарного состояния зависит только от скорости элиминации лекарства
- Повторное введение лекарства приводит к его существенному накоплению, если его принимают внутрь с интервалами, меньшими чем двойной конечный период полувыведения (2 Т1/2)
- Практически время установления стационарного состояния составляет 4-5 Т1/2
- Количество лекарства в организме при стационарном состоянии зависит от частоты приема и величины дозы
- Т1/2 лекарства в плазме не отражает метаболической способности, если изменяется условный объем распределения
Для быстрого создания терапевтической концентрации лекарства в плазме можно ввести ударную дозу (дозу насыщения)
Ударную дозу лекарства рассчитывают на основании его Vd и желаемой концентрации в плазме при стационарном состоянии (Css):
Ударная доза (мг/кг) = = Vd (л/кг) X Css (мг/л) (ф. 4.4)
При в/в введении ударную дозу обычно инфузируют в течение короткого периода времени, чтобы снизить риск возникновения побочных эффектов, связанных с очень высокой концентрацией лекарства в кровотоке. Затем рассчитывают величину поддерживающей дозы, основываясь на Сlр и величине интервалов между дозами (t):
Поддерживающая доза (мг/кг) = Сlр (л/кг/час) X Css (мг/л) X t (час) (ф. 4.5)
Источник


