- Фармакологические исследования, проводимые на животных
- Регуляторные и методические аспекты доклинических и клинических исследований кормовых добавок для животных
- Резюме
- Введение
- Международные регуляторные требования
- Доклинические исследования кормовых добавок для животных, проводимые на лабораторных животных
- Изучение острой токсичности
- Изучение субхронической токсичности
- Изучение хронической токсичности
- Изучение генотоксичности
- Изучение репродуктивной токсичности
- Исследование фармакокинетики кормовых добавок и остаточных веществ
- Исследования кормовых добавок для животных, проводимые на целевых животных
- Исследование переносимости кормовых добавок
Фармакологические исследования, проводимые на животных
Как указывалось выше, фармакологическое исследование новых препаратов включает фармакодинамическое и фармакокинетическое исследования. При проведении фармакодинамического исследования определяют специфическую активность препарата и побочные действия.
Для определения специфической активности применяют три следующих экспериментальных метода:
непосредственно прямую оценку действия исследуемого вещества на организм;
опосредованную непрямую оценку, заключающуюся в изучении особенностей воздействия исследуемого вещества на биохимические и биофизические процессы организма;
Прямой метод заключается в попытке вызвать у подопытного животного требуемый терапевтический эффект с помощью исследуемого вещества.
Непрямые методы не всегда достоверны, поскольку фармакологический эффект проявляется в разное время для патофизиологических процессов и биохимических и биофизических процессов.
Каждый лекарственный препарат имеет индивидуальные значения фармакокинетических показателей — объем распределения, время вывода половины вещества, клиренс.
В частности, объем распределения зависит от возможности проникновения через мембраны, способности соединяться с белками крови и тканей, от способности накапливаться в различных тканях.
Один и тот же лекарственный препарат может иметь разные значения фармакокинетических особенностей испытуемого организма (например, разные болезни или одинаковая болезнь, но в разных стадиях).
Этим можно объяснить разнообразие фармакологических и токсикологических данных для одного и того же лекарственного средства. Имеется непосредственная связь фармакодинамических и токсикологических эффектов в зависимости от исследуемого медикамента в плазме крови. Лекарственные препараты удаляются из организма либо в своем первоначальном виде, либо в виде метаболитов. Значимыми органами удаления лекарственных средств являются печень, почки и легкие. Биотрансформация лекарственного средства оказывает воздействие как на специфическую активность, так и на степень выраженности побочных эффектов.
Различие в биологическом эффекте связано с отличиями путей и скорости метаболических процессов. Именно этим определяется разная чувствительность к медицинским средствам отдельных представителей.
Токсикологические исследования включают следующие три основные группы:
исследование острой токсичности нового препарата D-l50;
исследование хронической токсичности нового вещества;
Исследование острой и хронической токсичности испытуемого препарата необходимо провести еще до проведения клинических испытаний. Данный аспект исследований позволяет выявить наиболее чувствительные к данному веществу ткани. Кроме того, это позволяет акцентировать внимание на необходимых моментах. Но при отсутствии явных токсических свойств данного препарата при проведении экспериментального исследования нельзя гарантировать его полную безопасность для человека.
В качестве подопытных животных применяются кролики, мыши и крысы, собаки. Токсикологическое исследование проводится одновременно на нескольких видах животных, так как возможны существенные видовые различия в чувствительности к новому препарату. По стандарту токсикологическое исследование испытуемого препарата известной фармакологической группы проводится не менее чем на двух видах животных. Для препарата новой группы исследования проводятся не менее чем на трех видах животных. При подведении итогов проведенного исследования необходимо учитывать возможное влияние условий содержания (микроклимата, питания, времени года).
При проведении испытаний выделяют две группы животных: контрольную и подопытную. При этом контрольная группа животных, в отличие от подопытной, получает индифферентное вещество.
Количество животных, взятых на исследование, должно быть приемлемым для объективной оценки частоты встречаемости токсического эффекта. Это составляет 15—50 особей для грызунов и 3—5 особей обоих полов для собак.
Результаты исследований подвергаются статистической обработке.
Для многокомпонентных препаратов токсичность каждого ингредиента проверяется отдельно, а затем только токсичность всей комбинации.
Рассмотрим более подробно отдельные этапы токсикологического исследования.
Исследование острой токсичности
При проведении исследований острой токсичности определяют величины летальных доз. В данном случае с помощью стандартных статистических методов определяется средняя смертельная доза — D-l50 (из 100 животных, участвующих в опыте, погибают 50).
Внимание исследователей привлекает определение максимально переносимой толерантной дозы и наблюдение симптомов интоксикации у погибших и выживающих животных. Все это дает полное представление о диапазоне терапевтического действия.
Продолжительность исследований острой токсичности от 3 суток до 2 недель. Важное значение имеет время наступления отравления и смерти подопытного животного. Быстрая гибель животных в течение 1 дня свидетельствует об активности препарата. Гибель на третьи сутки отражает повреждающее действие препарата на внутренние органы. Если определение смертельной дозы для данного вещества невозможно, то определяется максимальная доза, которая может быть введена животному без вреда для него.
Исследование хронической токсичности
Хроническая токсичность определяется при использовании препаратов в течение 1 года и более.
Данный эксперимент является наиболее трудоемким и дорогим. Так, по данным статистики стоимость одного такого исследования только для одного вещества составляет приблизительно 1000 долларов. В данном эксперименте определяются более чувствительные к препарату ткани, повреждающее действие нового лекарственного средства при длительном употреблении. Продолжительность проведения эксперимента зависит от курса лечения данным препаратом в стационаре, его кумулятивных свойств.
Исследование специфичной токсичности экспериментального препарата включает:
исследование воздействия на процесс воспроизводства потомства;2) исследование канцерогенности;
выявление индуцирования медикаментозной зависимости;
исследование аллергизирующих способностей;
Для поступления вещества в большой круг кровообращения оно должно пройти через биологические мембраны, печень. Для твердых форм веществ скорость всасывания зависит от процесса высвобождения лекарственного препарата непосредственно из лекарственной формы. Терапевтический эффект во многом обусловлен физическими свойствами лекарственного препарата (формой частиц, содержанием вспомогательных компонентов, влагосодержанием твердых лекарственных форм и т.д.). Так, вспомогательные ингредиенты способны самостоятельно влиять на организм.
При проведении клинических испытаний важным условием является возможность дополнительного производства препарата.
Для контроля качества лекарственных средств существует научно-техническая документация. Требования к лекарственным средствам различного происхождения разные. Научно-техническая документация необходима для гарантии соответствия выпускаемого медицинской промышленностью вещества препарату, который проходил клинические испытания. Для каждого нового лекарственного препарата проводятся доклинические, клинические и постклинические экспериментальные исследования фармакологических, фармацевтических и токсикологических характеристик. Требования к этим исследованиям непрестанно повышаются. Разрабатываются новые методические пособия для экспериментальных испытаний новых лекарств.
Источник
Регуляторные и методические аспекты доклинических и клинических исследований кормовых добавок для животных
Д.Р. Каргопольцева(1), ветеринарный врач группы специфической токсикологии, К.Л. Крышень(1), кандидат биологических наук, руководитель отдела токсикологии и микробиологии, М.Н. Макарова(2), доктор медицинских наук, директор, В.Г. Макаров(1), доктор медицинских наук, заместитель директора НПО «Дом Фармации» 1-Институт доклинических исследований, 188663, Россия, Ленинградская обл., Всеволожский район, г.п. Кузьмоловский, ул. Заводская, д. 3, к. 245; 2-Научно-производственное объединение «Дом Фармации», 188663, Россия, Ленинградская обл., Всеволожский район, г.п. Кузьмоловский, ул. Заводская, д. 3, к. 245 Е-mail: agasieva.dr@doclinika.ru
Резюме
В последние годы в связи с развитием сельского хозяйства и ростом числа владельцев домашних животных происходит стремительное развитие ветеринарии не только в Российской Федерации, но и во всем мире. Попутно с развитием ветеринарной медицины идет и совершенствование ветеринарной фармацевтики, которая разрабатывает и производит все больше ветеринарных препаратов и кормовых добавок для животных. Для попадания на рынок новых кормовых добавок для животных, так же как и лекарственных препаратов для человека, требуется государственная регистрация с соответствующим пакетом документов. Наиболее важные документы – результаты доклинических и клинических исследований. Однако если в отношении лекарственных препаратов для человека в Российской Федерации существует множество нормативных документов, которые описывают объем и схему проведения исследований, то в отношении кормовых добавок таковых документов и инструкций до настоящего времени не разработано. Поэтому требуется разработка регламентирующих документов, гармонизированных с международными требованиями. В данной работе рассмотрены основные международные регуляторные требования к проведению доклинических и клинических исследований кормовых добавок для животных.
Введение
В последние годы в Российской Федерации особое внимание уделяется развитию и совершенствованию сельского хозяйства. Сельское хозяйство – одна из ключевых и фундаментальных отраслей экономики любого государства, направленная на обеспечение населения продуктами питания и получение сырья для ряда областей промышленности. Одной из отраслей сельского хозяйства является животноводство, успех развития которого зависит от здоровья и продуктивности сельскохозяйственных животных. В связи с этим наблюдается непрерывное расширение ассортимента кормовых добавок для животных, которые не всегда отличаются хорошим качеством.
В п. 4 проекта Административного регламента предоставления Россель-хознадзором государственной услуги по государственной регистрации кормовых добавок приведено следующее определение: «кормовые добавки – это продукты или их комбинации растительного, животного, микробиологического, минерального и синтетического происхождения, предназначенные для включения в состав кормов и рационов животных с целью обеспечения физиологической полноценности, стимуляции продуктивности животных, обеспечения сохранности компонентов, увеличения доступности питательных веществ, улучшения вкусовых и технологических свойств кормов» [1].
Использование некачественных кормовых добавок может негативно сказаться не только на самих сельскохозяйственных животных, но и на здоровье людей, потребляющих продукцию, получаемую от этих животных.
Для поступления кормовой добавки в ветеринарную аптеку необходимо пройти государственную регистрацию, которую осуществляет Россельхознадзор на базе государственного учреждения «Всероссийский государственный центр контроля качества и стандартизации лекарственных средств для животных и кормов» (ФГУ «ВГНКИ») в течение 6 мес со дня подачи регистрационных документов. В соответствии с приказом Минсельхоза РФ №48 от 1 апреля 2005 г. «Об утверждении Правил государственной регистрации лекарственных средств для животных и кормовых добавок» [2] обязательной регистрации подлежат:
- новые добавки;
- новые комбинации зарегистрированных ранее добавок;
- добавки, зарегистрированные ранее, но произведенные в других формах, или с новой дозировкой, или с другим составом вспомогательных веществ;
- воспроизведенные добавки.
Для государственной регистрации кормовых добавок в Россельхознадзор необходимо предоставлять результаты доклинических исследований. Проведение доклинических исследований является обязательной процедурой для регистрации кормовой добавки и занесения ее в Государственный реестр кормовых добавок.
Таким образом, производитель должен составить программу и провести доклинические исследования разрабатываемых кормовых добавок для животных с целью изучения их эффективности, безопасности применения для здоровья животных и подтверждения их качества.
Как и в каком объеме проводить доклинические исследования кормовых добавок для животных? Для медицинских препаратов давно существуют руководства, стандартные правила и методические указания по организации и проведению доклинических исследований. В отношении ветеринарных препаратов до недавнего времени такие правила отсутствовали. Сегодня разработан Приказ Министерства сельского хозяйства Российской Федерации №101 от 06.03.2018 «Об утверждении правил проведения доклинического исследования лекарственного средства для ветеринарного применения, клинического исследования лекарственного препарата для ветеринарного применения, исследования биоэквивалентности лекарственного препарата для ветеринарного применения» [3]. В то же время в отношении кормовых добавок для животных правил по проведению исследований до настоящего времени в Российской Федерации не разработано. В связи с этим целью данной публикации стало рассмотрение международных регламентирующих документов.
Международные регуляторные требования
При рассмотрении вопроса о кормовых добавках в Европейских странах руководствуются Регламентом комиссии (ЕС) №1831/2003 [4], который описывает порядок выдачи разрешений на размещение на рынке и применение кормовых добавок, правил контроля и маркировки кормовых добавок и премиксов в качестве основы для обеспечения высокого уровня защиты здоровья человека, здоровья и благополучия животных, окружающей среды и интересов потребителей, а также обеспечение эффективного функционирования внутреннего рынка. При проведении исследований токсичности и эффективности кормовых добавок руководствуются Регламентом №429/2008 [5], в котором содержатся правила по подготовке и представлению заявок, а также проведению оценки и выдаче разрешений в отношении кормовых добавок.
В соответствии с Регламентом (ЕС) №1831/2003 [4], если дело касается кормовых добавок, то исследования токсичности и эффективности не требуются в отношении:
- мочевины;
- аминокислот, их солей и аналогов;
- микроэлементов и витаминов, провитаминов и веществ с установленным химическим составом, имеющих схожее действие и не накапливающихся в организме, уже разрешенных к применению в виде кормовых добавок;
- кормовых добавок, произведенных путем ферментации, в случае если производственный организм рассматривается европейским агентством по безопасности пищевых продуктов в качестве организма с установленным безопасным статусом;
- кормовых добавок, произведенных путем ферментации, действующее вещество которых отделяется от необработанного ферментированного продукта и подвергается высокой очистке.
Токсикологические исследования и исследования эффективности проводятся в целях совершенствования состава:
- витаминов, провитаминов и веществ с установленным химическим составом, имеющих схожее действие и способных к накоплению, предполагаемая или подтвержденная способность к накоплению которых отличается от способности к накоплению витамина(-ов) с установленным составом;
- производных мочевины;
- аналогов аминокислот и соединений микроэлементов ранее не одобренных;
- кормовых добавок, произведенных путем ферментации, не исключенных по вышеописанным правилам.
Исследования токсичности и эффективности кормовых добавок для животных проводят на лабораторных животных (проявляющих наибольшую чувствительность) и на целевых видах животных (табл. 1). Такие исследования осуществляют в соответствии с Директивой 2010/63/EU Европейского Парламента и Совета Европейского Союза по охране животных, используемых в научных целях, от 22.09.2010 [6].
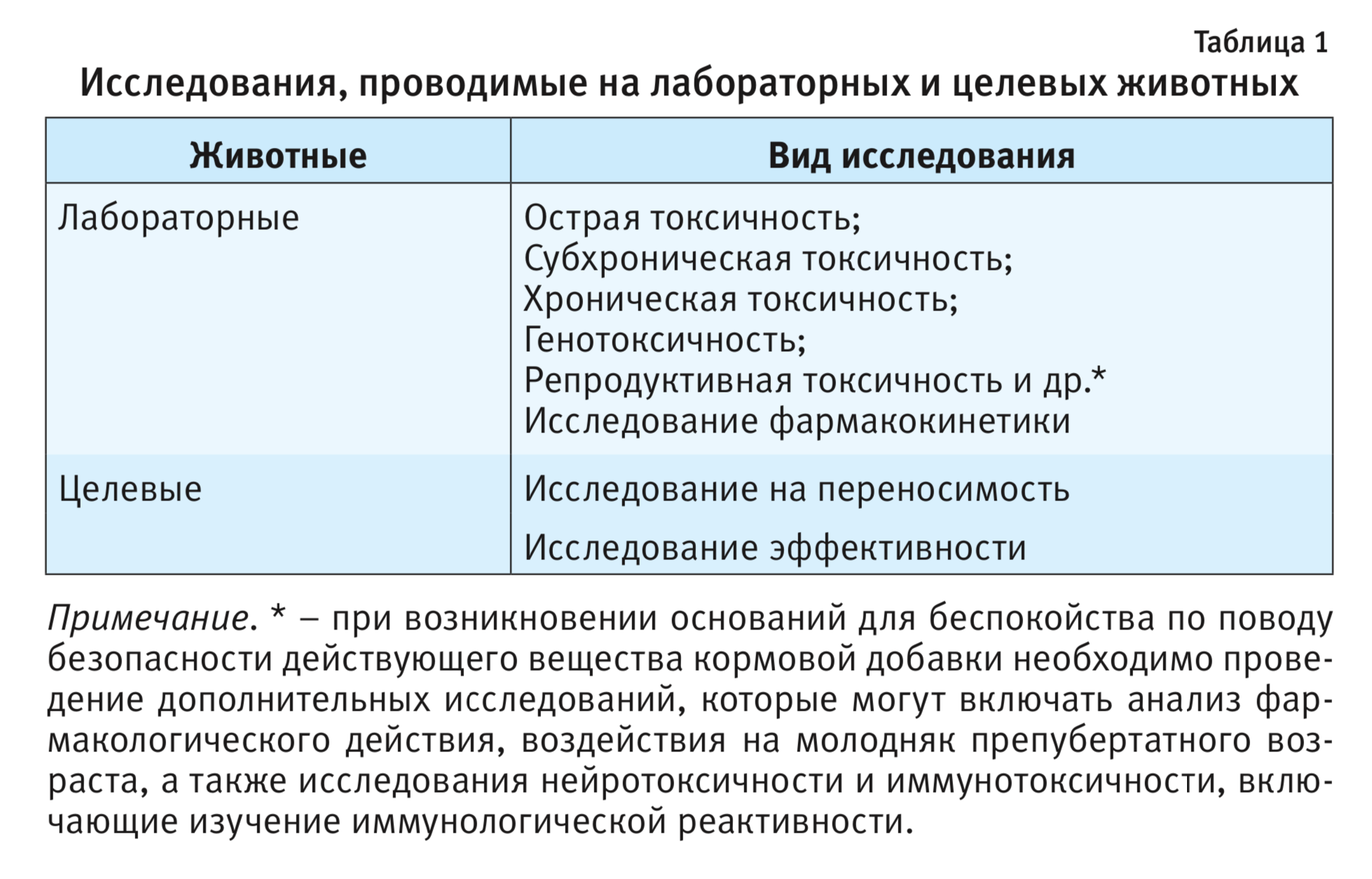
Далее рассмотрим необходимые исследования, проводимые на лабораторных и целевых животных.
Доклинические исследования кормовых добавок для животных, проводимые на лабораторных животных
Изучение острой токсичности
Исследования острой токсичности проводятся с целью установления принадлежности тестируемого объекта к определенному классу токсичности. Исследования острой токсичности осуществляются не менее чем на 2 видах млекопитающих. В случае необходимости один вид лабораторных животных может быть заменен целевым видом. Точное определение летальной дозы LD50 не является необходимостью, достаточным считается определение приблизительного значения летальной дозы [4, 5]. При проведении токсикологических исследований по изучению острой токсичности следует соблюдать Руководящие принципы ОЭСР (OECD Test No.420 «Acute Oral Toxicity – Fixed Dose Procedure», ГОСТ 32296-2013; Test No.423 «Acute Oral Toxicity – Acute Toxic Class Method», ГОСТ 32644-2014) [7].
Изучение субхронической токсичности
Изучение субхронической токсичности тестируемых объектов проводят на мышах и крысах при пероральном/внутрижелудочном введении в течение не менее 90 сут. При необходимости исследование субхронической токсичности дополнительно проводится на животных, не относящихся к отряду грызунов. Для установления дозозависимых эффектов тестируемых объектов в исследование субхронической токсичности должно быть включено не менее 3 уровней доз, при этом минимальная доза не должна оказывать токсического действия на организм. Также должна быть сформирована контрольная группа, с помощью которой оцениваются результаты проведенного экспериментального исследования [4, 5]. При проведении исследований по изучению субхронической токсичности следует соблюдать Руководящие принципы ОЭСР (OECD Test No.408 «Repeated Dose 90-Day Oral Toxicity Study in Rodents», ГОСТ 32637-2014; Test No.409 «Repeated Dose 90-Day Oral Toxicity Study in Non-Rodents», ГОСТ Р 56697-2015) [7].
Изучение хронической токсичности
Для установления хронической токсичности исследования тестируемых объектов выполняют на протяжении не менее 12 мес. Так же, как при изучении субхронической токсичности, изучение хронической токсичности происходит с исследованием как минимум 3 доз и с добавлением контрольной группы животных [4, 5]. При проведении исследований по изучению хронической токсичности следует соблюдать Руководящие принципы ОЭСР (OECD Test No.452 «Chronic Toxicity Studies», ГОСТ 32437-2013; Test No.453 «Combined Chronic Toxicity/Carcinogenicity Studies», ГОСТ 32647-2014) [7].
В документе Регламента комиссии (ЕС) № 429/2008 не предусмотрено более подробное рассмотрение вопроса о длительности проведения исследования хронической токсичности, а также возможности проведения исследования хронической токсичности без исследования субхронической токсичности. Такие длительные исследования очень дорогостоящие и трудозатратные. Заметим, что доклинические исследования лекарственных препаратов для человека регламентируются ГОСТ Р 56701-2015 [8], где рекомендуемая продолжительность исследования общетоксических свойств при многократном применении у животных соответствует продолжительности применения препарата в клинике (табл. 2).
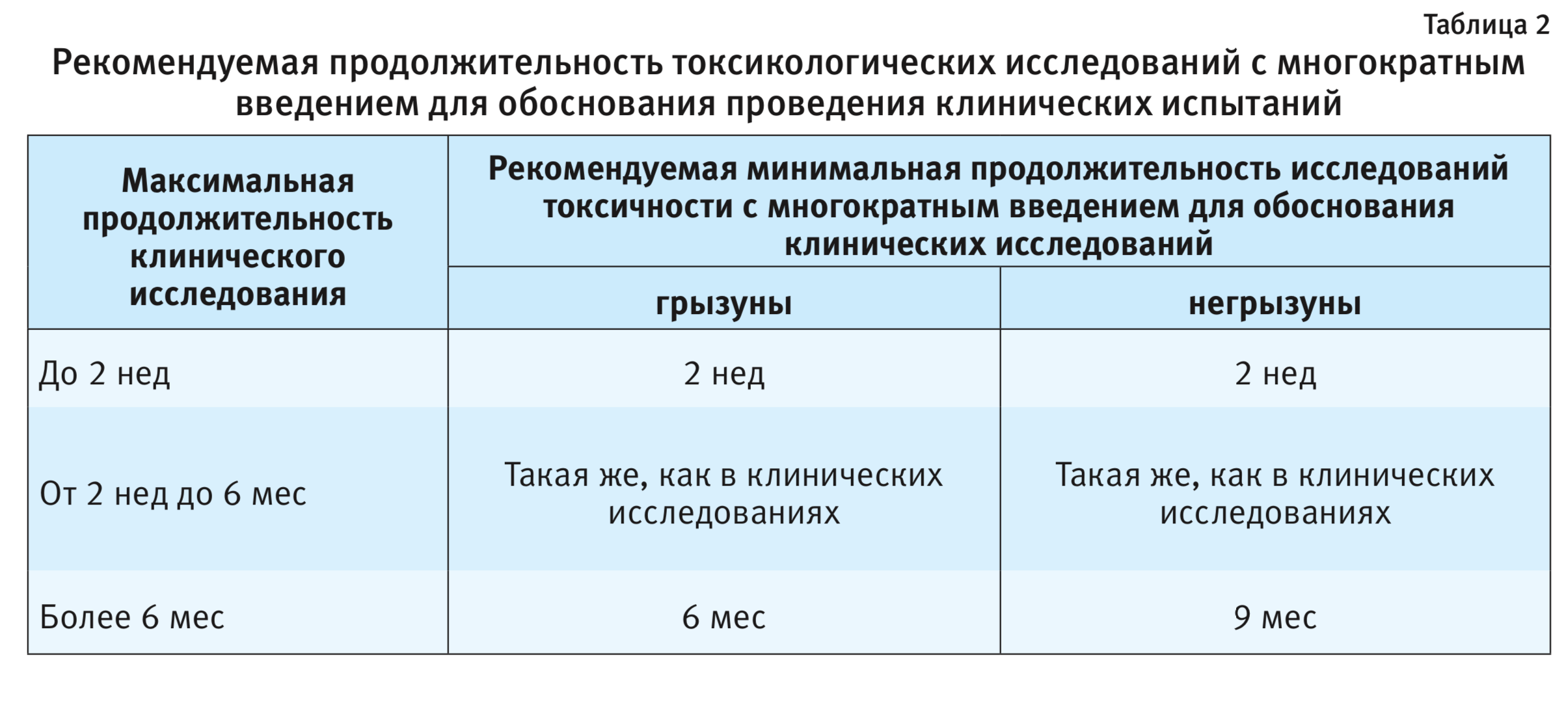
Изучение генотоксичности
Для изучения генотоксичности тестируемых объектов существует целый ряд испытаний:
- тесты на индукцию генных мутаций у бактерий и в клетках млекопитающих;
- индукция хромосомных аберраций в клетках млекопитающих;
- испытания in vivo.
При проведении исследований по изучению генотоксичности следует соблюдать Руководящие принципы ОЭСР (OECD Test No.471 «Bacterial Reverse Mutation Test», ГОСТ 32376-2013, Test No.473 «In Vitro Mammalian Chromosomal Aberration Test»; Test No.474 «Mammalian Erythrocyte Micronucleus Test» ГОСТ 32635-2014; Test No.476 «Mammalian Bone Marrow Chromosomal Aberration Test»; Test No.476 «In Vitro Mammalian Cell Gene Mutation Tests using the Hprt and xprt
genes») [7].
В регламенте Комиссии (ЕС) №429/2008 нет четких рекомендаций по объему исследований генотоксичности кормовых добавок для животных. Если рассматривать исследования генотоксичности лекарственных препаратов для человека, то существуют рекомендации ГОСТ 57130-2016 [9], где предлагается проведение следующей батареи тестов:
Вариант 1
- Испытание генных мутаций в бактериях.
- Цитогенетический тест хромосомных повреждений (анализ хромосомных аберраций в метафазе invitro или микроядерная проба in vitro) либо анализ генных мутаций Tk в лимфоме мышей in vitro.
- Тест генотоксичности invivo, обычно тест на хромосомные поломки на гемопоэтических клетках мышей в виде либо микроядерной пробы либо анализа на хромосомные аберрации в метафазе.
Вариант 2
- Испытание генных мутаций в бактериях.
- Оценка генотоксичности invivo двух разных тканей, обычно микроядерная проба с использованием гемопоэтических клеток грызунов и 2-й анализ in vivo. Как правило, используется анализ поломки нитей ДНК в печени.
Изучение репродуктивной токсичности
Цель изучения репродуктивной токсичности – установление возможных неблагоприятных воздействий тестируемого объекта на мужскую и женскую репродуктивную систему, а также патологического воздействия на развитие потомства. При проведении исследований репродуктивной токсичности тестируемый объект вводят в течение определенного времени до ссадки самцов и самок. Затем наблюдают за эффективностью оплодотворения, течением беременности и родов у самок, далее контролируют рост и развитие потомства [4, 5]. При проведении исследований по изучению репродуктивной токсичности следует соблюдать Руководящие принципы ОЭСР (OECD Test No.416 «Two-Generation Reproduction Toxicity Study», ГОСТ Р 56698-2015) [7].
Также изучение репродуктивной токсичности включает в себя исследование токсических эффектов на внутриутробное развитие потомства, целью которого является установление неблагоприятного воздействия тестируемого объекта на беременных самок и плод при введении объекта на протяжении всего периода беременности [5, 6]. В ходе исследований следует соблюдать Руководящие принципы ОЭСР (OECD Test No.414 «Prenatal Development Toxicity Study», ГОСТ 32380-2013) [7].
В документе Регламента комиссии (ЕС) № 429/2008 также нет достаточно подробного описания необходимости проведения исследования тестируемых объектов на беременных самках. По нашему мнению, данной необходимости нет, если нет опасений в неблагоприятном воздействии кормовой добавки на течение беременности и плод в клинике и, если кормовая добавка не предусмотрена к применению в период беременности.
Исследование фармакокинетики кормовых добавок и остаточных веществ
Решающим фактором при идентификации и количественном определении остаточных веществ в продуктах питания, полученных от животных (молоко, мясо, субпродукты и др.), в рацион которых была включена кормовая добавка, является установление метаболического пути добавки у целевых видов животных. При исследовании фармакокинетики представляются результаты исследований всасывания, распределения, метаболизма и экскреции (выведения) действующего вещества (и его метаболитов) кормовой добавки.
Исследования фармакокинетики действующего вещества кормовых добавок должны проводиться при введении животным в максимальной дозе, согласно инструкции по применению кормовой добавки. Исследования предусматривают приблизительную оценку скорости и степени всасывания, распределения в крови и выделения с экскретами (с мочой, желчью, экскрементами, молоком, яйцами), а также установление фармакокинетического профиля, порядка распределения действующего вещества в тканях и продуктах после введения повторных доз до достижения метаболического равновесия.
Исследования должны включать в себя идентификацию остаточных веществ действующего вещества, оказывающих токсическое действие, в продуктах животноводства (печень, почки, мышцы, кожа, кожа + жир, молоко, яйца), а также установление периода выведения.
Фармакокинетические исследования проводятся на лабораторных видах животных. Однако в случае исследований на определенные метаболиты могут потребоваться дополнительные исследования, если такие метаболиты вырабатываются в организме целевых животных и не образуются в значительной степени в организме лабораторных животных.
Исследования кормовых добавок для животных, проводимые на целевых животных
Согласно Регламентам (ЕС) №1831/2003 [4] и №429/2008 [5], исследования проводятся также на целевых видах животных, которые включают в себя выявление переносимости и эффективности кормовых добавок.
Исследование переносимости кормовых добавок
При установлении переносимости кормовой добавки проводят кратковременные исследования, длительность которых зависит от вида животных (табл. 3–7). Обычно исследуют максимально рекомендуемую дозу и дозы в 10 раз ее превышающие, при этом учитывают результаты доклинических исследований на лабораторных животных. В случае переносимости кормовых добавок целевыми видами животных в дозах, превышающих максимально рекомендуемые менее чем в 10 раз, при проведении исследования необходимо предусматривать определение предельно допустимого безопасного уровня добавки.
Источник


