- Исследование терапевтической эквивалентности лекарственных препаратов это
- Исследование терапевтической эквивалентности лекарственных препаратов это
- Что такое дженерик (воспроизведенный препарат)
- Значимость исследований по терапевтической эквивалентности
- «Дженериковая замена»
- На что должен ориентироваться практический врач при выборе воспроизведенных препаратов
Исследование терапевтической эквивалентности лекарственных препаратов это

Российский Федеральный закон «Об обращении лекарственных средств» вводит понятие воспроизведенного лекарственного средства, однако входит в некоторое противоречие с документами других стран. В соответствии с Федеральным законом Российской Федерации от 12 апреля 2010 г. N 61-ФЗ» при проведении процедуры экспертизы воспроизведенных лекарственных средств должна быть представлена информация, полученная при проведении клинических исследований лекарственных препаратов и опубликованная в специализированных печатных изданиях, а также документы, содержащие результаты исследования биоэквивалентности и (или) терапевтической эквивалентности. Если говорить об исследованиях терапевтической эквивалентности лекарственных препаратов, то под этим термином понимается достижение клинически сопоставимого терапевтического эффекта при применении лекарственных препаратов для медицинского применения для одной и той же группы больных по одним и тем же показаниям к применению [1, 2].
Несмотрям на важность показателей биоэквивалнтности при регистрации воспроизведенного лекарственного препарата, результаты клинических исследований для доказательства терапевтической эквивалентности сохраняют определенную значимость.
Есть данные, которые подтверждают отсутствие терапевтической эквивалентности (ТЭ) лекарственных препаратов (ЛП) при доказанной биоэквивалентности. Так, при иследовании клинической эффективности четырех воспроизведенных препаратов эналаприла с референтным препаратом они оказались терапевтически неэквивалентны при доказанной фармацевтической и биоэквивалентности [3].
Если говорить об антиаритмических препаратах (ААП), то эта группа препаратов обладает узким терапевтическим диндексом [4], что по данным FDA является фактором определяющим проведение оценки ТЭ.
Современные представления об эффективности и безопасности лечения антиаритмическими препаратами базируются в первую очередь на сведениях о влиянии препаратов на так называемые «конечные точки»: общую смертность и внезапную смерть пациентов. В работе F.T. McAlister и K.K. Teo [5] изучены результаты многочисленных исследований, посвященных антиаритмической терапии. Авторы приходят к следующим выводам:
– профилактическое назначение препаратов I класса (мембраностабилизаторов) у больных высокого риска, преимущественно перенесших инфаркт миокарда (ИМ) связано с достоверным повышением риска смерти (61 исследование, 23486 больных);
– препараты II класса, бета-адреноблокаторы, значительно снижают риск смерти у больных после ИМ (56 исследований, 53521 больной);
– данные о лечении больных высокого риска (после ИМ миокарда, с сердечной недостаточностью [СН по классификации NYHY], переживших остановку сердца) препаратом III класса амиодароном подтверждают его эффективность в снижении риска смерти (14 исследований, 5713 больных);
– препараты IV класса, блокаторы медленных кальциевых каналов, не снижают у пациентов риск внезапной смерти (26 исследований, 21644 больных).

Рис. 1. Изменение сывороточной концентрации амиодарона (мг/л) после замены Кордарона на дженерик у 28-летнего мужчины с желудочковой тахикардией
Из этих данных следует, что лишь препараты, обладающие свойствами антиаритмиков II и III класса, снижают риск внезапной смерти, то есть имеют как антиаритмическую, так и антифибрилляторную активность. Подтверждением этому могут служить результаты большого числа многоцентровых исследований, таких, как MIAMI, GMT, SMT, BASIS, EMIAT, EPASMA, SSSD.
Далее мы приводим ряд клинических исследований воспроизведенных препаратов амиодарона, который является «золотым стандартом» антиаритмических лекарственных препаратов.
После появления первых дженериков амиодарона в литературе были опубликованы сообщения о фармакологических и клинических аспектах замены оригинального препарата на его копии. S. Sauro и соавт. [6] сопоставили равновесные концентрации амиодарона и дезэтиламиодарона у 138 пациентов, принимавших Кордарон в стабильной дозе, а затем перешедших на лечение дженериком амиодарона. Равновесные уровни амиодарона и его метаболита достоверно не изменились после замены оригинального препарата дженериком, однако вариабельность концентрации препарата в плазме увеличилась. По мнению авторов, в течение 1-3 месяцев после смены препарата целесообразно контролировать концентрации действующего вещества в плазме (в России эта рекомендация практически не выполнима).
J. Reiffel и P. Kowey [7] провели опрос 130 ведущих американских аритмологов, которым предлагали сообщить, наблюдали ли они рецидивы аритмий при замене оригинальных антиаритмических препаратов на дженерики. На поставленные вопросы ответили 64 специалиста. Около половины из них наблюдали эпизоды аритмий (включая фибрилляцию желудочков, желудочковую тахикардию, фибрилляцию предсердий и предсерную тахикардию), которые были определенно или вероятно связаны с заменой оригинального препарата. В целом было зарегистрировано 54 рецидива аритмий, включая 32 случая при замене Кордарона на дженерик амиодарона. Три пациента умерли (в том числе пациент, получавший амиодарон). Кроме того, эксперты наблюдали 7 случаев аритмогенного действия дженериков (один из них был зарегистрирован при применении дженерика амиодарона, хотя этот препарат характеризуется низкой аритмогенной активностью). В части случаев связь между рецидивами аритмий и заменой антиаритмического препарата была подтверждена при повторной провокации или анализе сывороточных уровней лекарственных веществ в плазме (рис. 1).
Таким образом, около половины респондентов сталкивались с проблемами при смене антиаритмического препарата, причем во всех этих случаях оригинальный препарат заменяли на его копию.
P. Pollak [8] определял концентрации амиодарона и его активного метаболита десэтиламиодарона у 77 пациентов, длительно принимавших Кордарон. После начала лечения отношение концентраций метаболита и амиодарона увеличивалось примерно до 0,9 и не зависело от дозы препарата. Этот показатель редко превышал 1,4, а его увеличение ассоциировалось с токсическими изменениями со стороны легких. У 4 пациентов замена оригинального Кордарона на дженерик сопровождалась значительным увеличением отношения уровней дезэтиламиодарона и амиодарона (рис. 2). Например, у 42-летнего мужчины, принимавшего Кордарона по поводу предсердной тахикардии, через 1 месяц после назначения дженерика указанный коэффициент увеличился с 0,8 до 1,8. После возобновления приема Кордарона он снизился до 1,1. Еще одна попытка замены оригинального препарата вновь привела к увеличению отношения уровней метаболита амиодарона до 1,6. Следует отметить, что во всех 4 случаях относительные изменения концентраций метаболита существенно превышали 20 % (максимальный показатель для биоэквивалентного дженерика). Для подтверждения биоэквивалентности дженериков амиодарона не требуется анализ активного метаболита этого препарата, хотя изменения его уровня могут отражаться на эффективности и безопасности лечения.
Сходные проблемы наблюдались и при применении других антиаритмических препаратов. B. Grubb [9] описал рецидив желудочковой тахикардии после замены оригинального прокаинамида на дженерик. При обследовании было выявлено снижение сывороточной концентрации действующего вещества до 2,4 мг/мл (эффективный уровень – около 10 мг/мл). Т. Ozahowski и соавт. [10] наблюдали 2 случая рецидивирующей наджелудочковой тахикардии у пациентов с имплантированным кардиовертером-дефибриллятором, получавших дженерик прокаинамида замедленного высвобождения. Контроль аритмии был восстановления после возобновления приема оригинального препарата. В описанном выше исследовании J. Reiffel и P. Kowey [11], которые проводили опрос американских аритмологов, рецидивы аритмий или аритмогенные эффекты отмечались при смене препаратов не только амиодарона, но и прокаинамида, хинидина, дизопирамида.
Приведенные данные могут показаться не очень убедительными. На самом деле они представляют собой описания отдельных случаев, а не результаты рандомизированных контролируемых исследований. Однако известно, что именно постмаркетинговое наблюдение позволяет выявить серьезные нежелательные последствия или взаимодействия лекарственных средств, которые могут быть даже причиной прекращения их маркетинга (примерами могут служить антигистаминные препараты терфенадин и астемизол, статин церивастатин и др.). Последнее обычно происходит, если имеются более безопасные представители того же класса. Распознать нежелательные эффекты замены оригинального антиаритмического препарата на дженерики в клинической практике очень сложно, особенно при отсутствии четкой хронологической зависимости [12].
Врачи обычно не придают особого значения тому, какой препарат применяется – оригинальный или воспроизведенный. При этом рецидив аритмии, скорее всего, будет расценен как следствие прогрессирования основного заболевания, а не смены препарата. Подтверждением этой связи могут быть изменения концентрации действующего вещества и/или его метаболитов в крови, однако на практике врачу проще отменить соответствующий препарат, чем провести эти исследования. J. Reiffel [13] предложил следующие рекомендации по замене оригинальных антиаритмических препаратов на воспроизведенные:
— заменять антиаритмические препараты не следует у пациентов с угрожающими жизни аритмиями, аритмиями, которые могут вызвать потерю сознания, а также в тех случаях, когда повышение уровней лекарственного вещества в крови может привести к аритмогенному действию;
– не следует заменять антиаритмические препараты, биотрансформирующиеся до множественных метаболитов или метаболитов, которые нельзя определить;
– при менее серьезных аритмиях дженерики можно применять только в тех случаях, когда имеется простой и надежный метод мониторирования их концентрации;
– при необходимости замены следует тщательно контролировать уровни препарата в крови.
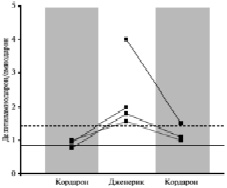
Рис. 2. Изменения отношения уровней дэзэтиламиодарона/амиодарона при замене Кордарона на джененики. Сплошная линия – средний показатель у 77 пациентов, пунктирная – 95-й перцентиль
Если снижение или повышение концентрации антиаритмического препарата может привести к угрожающим жизни последствиям, замена оригинального препарата возможно только в следующихслучаях:
– зарегистрирован только один воспроизведенный препарат и, соответственно, отсутствует риск многочисленных замен;
– воспроизведенный препарат широко доступен в стационарах и аптечной сети.
Если выполнять все эти рекомендации, то от применения воспроизведенных антиаритмических препаратов пришлось бы полностью отказаться как при легких (из-за невозможности мониторирования концентраций в крови), так и серьезных нарушениях ритма. На практике следует, вероятно, придерживаться следующей тактики ведения. Если пациент получает антиаритмическую терапию оригинальным препаратом с хорошим эффектом, то заменять его на воспроизведенный препарат не следует. Если по экономическим причинам замена оригинального антиаритмического препарата все же необходима, то пациент должен находиться под наблюдением, чтобы убедиться в сохранении достигнутого ранее эффекта. При рецидиве аритмии или ухудшении переносимости можно попытаться возобновить прежнюю терапию. В любом случае целесообразно избегать частых замен копий оригинального препарата. Единственным основанием для замены оригинального препарата на воспроизведенный являются экономические соображения. Соответственно, возникает вопрос – какова экономия затрат при замене Кордарона на дженерики? Розничная стоимость упаковки Кордарона (30 таблеток по 200 мг), которой достаточно на месяц поддерживающей терапии, составляет около 250 рублей. Стоимость дженериков примерно в 2-3 раза ниже. Следовательно, экономия составит не более 150-180 рублей в месяц. Вряд ли, указанная сумма оправдывает возможные последствия подобной модификации антиаритмической терапии. Необходимо учитывать, что ухудшение эффективности и переносимости лечения приводит к росту затрат (госпитализации, дополнительные визиты к врачу, вызов скорой помощи и т.п.), поэтому в конечном итоге общая стоимость лечения может даже увеличиться [12]. По мнению P. Pollak [8], если только 5 % больных, получающих амиодарон в США и Канаде, потребуется одна госпитализация, связанная с заменой оригинального препарата на воспроизведенный, то общее число таких госпитализаций составит 20000 в год.
На примере амиодарона очевидно, что проведение качественных исследований терапевтической эквивалентности воспроизведенных антиаритмических лекарственных препаратов очень важно для обеспечения эффективности, безопасности, экономической выгоды применения антиаритмических лекарственных препартов, а в ряде случаев может спасти жизнь пациента.
Источник
Исследование терапевтической эквивалентности лекарственных препаратов это
Терапевтическая эквивалентность воспроизведенного препарата (дженерика) и как ее доказать.
Н.П.Кутишенко1, С.Ю.Марцевич1,2, И.В.Вашурина1
1ФГУ ГНИЦ ПМ Минздравсоцразвития России, Москва
2Кафедра доказательной медицины Первого МГМУ им. И.М.Сеченова
Проблема эффективности и безопасности препаратов-копий (воспроизведенных препаратов, дженериков) продолжает беспокоить ученых, врачей, общественность. К ней постоянно обращаются на научных конференциях и симпозиумах, в средствах массовой информации [1, 2], ей посвящаются специальные научные исследования, в которых иногда участвуют тысячи больных, например, исследование ОРИГИНАЛ (Оценка эффективности пеРевода с Индапамидов ГенерИков На Арифон ретард у пациентов с АртериаЛьной гипертензией) [3]. И все это несмотря на то, что научная часть этой проблемы в основном уже давно решена в многочисленных исследованиях, а ее практическая часть нашла отражение в целом ряде нормативных документов, о которых речь пойдет ниже. Характерно, что в зарубежной научной литературе сейчас крайне редко появляются публикации, посвященные сравнительной оценке оригинальных препаратов и дженериков, хотя совсем недавно таких публикаций было значительно больше [4].
Безусловно, определенные неясности в отношении эффективности и безопасности некоторых дженериков остаются, однако они, по нашему мнению, отражают в первую очередь проблемы с соблюдением тех необходимых условий, которые, по современным представлениям, обеспечивают терапевтическую эквивалентность препарата-дженерика.
Цель данной публикации как раз и заключается в том, чтобы напомнить об основных принципах оценки терапевтической эквивалентности воспроизведенных лекарственных препаратов.
Что такое дженерик (воспроизведенный препарат)
Как это ни покажется странным, но единого определения понятия «дженерик» до сих пор не существует: ВОЗ (Всемирная Организация Здравоохранения), FDA (Food and Drug Administration), EMEA (European Medicines Agency), министерства здравоохранения различных стран предлагают свои определения для воспроизведенного препарата, а также критерии, на основании которых дженерик можно считать терапевтически эквивалентным оригинальному препарату. В целом эти критерии совпадают, однако есть определенные различия в оценке значимости и необходимости проведения исследований по терапевтической эквивалентности для доказательства соответствия дженерика оригинальному препарату как по эффективности, так и по безопасности.
Без сомнения, наиболее четкая, продуманная и научно обоснованная система оценки эквивалентности дженериков на сегодняшний день существует в США, которая отражена в документах FDA. В соответствии с определением FDA, терапевтическая эквивалентность устанавливается в ходе исследований фармацевтической эквивалентности и биоэквивалентности. Если сомнений в эквивалентности нет, то препарату присваивается соответствующий код, начинающийся с буквы «А», что также означает, что он может рассматриваться как возможный референсный препарат (т.е. препарат сравнения). Если данные биоэквивалентности не исключают потенциальных сомнений в отношении терапевтической эквивалентности фармацевтически эквивалентных препаратов или изучение биоэквивалентности не проводилось (н-р, для препаратов местного действия), то код оценки терапевтической эквивалентности начинается с буквы «В». Большинство дженериков в соответствии с данной системой кодировки, как правило, получают код «АВ» – это означает, что различия между препаратами потенциально возможны, но эквивалентность подтверждается результатами адекватно выполненных in vitro или/и in vivo исследований. Следует отметить, что проведение специальных клинических исследований, подтверждающих терапевтическую эквивалентность оригинального препарата и дженерика, не предполагается [5].
ВОЗ определяет терапевтическую эквивалентность оригинального препарата и дженерика (мультиисточникового фармацевтического продукта) несколько иначе. В соответствии с требованиями ВОЗ два фармацевтических продукта считаются терапевтически эквивалентными в том случае, если они фармацевтически эквивалентны (или являются фармацевтически альтернативными) и после введения в одинаковой молярной дозе их эффект в отношении эффективности и безопасности совершенно одинаков при одинаковом способе назначения и при одинаковых показаниях. Это должно быть продемонстрировано с помощью соответствующих исследований биоэквивалентности, таких как фармакокинетические, фармакодинамические, клинические или в исследованиях in vitro [6].
С точки зрения EMEA (European Medicines Agency) исследования по биоэквивалентности необходимы не только для того, чтобы продемонстрировать сходство между дженериком и оригинальным препаратом по основным фармакокинетическим показателям. Такие исследования предоставляют реальную возможность переноса данных об эффективности и безопасности, полученных для оригинального препарата, на дженерик, при этом проведение исследований терапевтической эквивалентности не предполагается (исключение – биологические лекарственные препараты) [7].
Российский Федеральный закон «Об обращении лекарственных средств» вводит понятие воспроизведенного лекарственного средства, однако входит в некоторое противоречие с документами других стран. В соответствии с Федеральным законом Российской Федерации от 12 апреля 2010 г. N 61-ФЗ » при проведении процедуры экспертизы воспроизведенных лекарственных средств (к ним как раз и относятся дженерики) должна быть представлена информация, полученная при проведении клинических исследований лекарственных препаратов и опубликованная в специализированных печатных изданиях, а также документы, содержащие результаты исследования биоэквивалентности и (или) терапевтической эквивалентности. Если говорить об исследованиях терапевтической эквивалентности лекарственных препаратов, то под этим термином понимается вид клинического исследования, проведение которого осуществляется для выявления одинаковых свойств лекарственных препаратов определенной лекарственной формы, а также наличия одинаковых показателей безопасности и эффективности лекарственных препаратов, одинаковых клинических эффектов при их применении [8].
Относительно вопроса о подтверждении терапевтической эквивалентности сложились определенные противоречия с правилами FDA, также отсутствуют документы, определяющие порядок проведения и критерии оценки результатов таких клинических испытаний. Если обратиться к проверенным временем правилам FDA по определению терапевтической эквивалентности, то для этого должны быть в обязательном порядке соблюдены пять условий: 1) препараты должны быть признаны эффективными и безопасными, 2) они должны быть фармацевтически эквивалентными, включая соответствие по количеству активных ингредиентов, их чистоте, качеству, идентичности, 3) соответствовать стандартам биоэквивалентностии при участии в исследовании не менее 24-36 добровольцев, 4) правильно промаркированы и, что не менее важно, 5) произведены в соответствии с требованиями GMP (Good Manufacturing Practice) [5].
Значимость исследований по терапевтической эквивалентности
Однако, несмотря важность показателей биоэквивалнтности при регистрации дженерика, результаты клинических исследований для доказательства эквивалентности сохраняют определенную значимость. В большей мере это касается аналогов фармацевтических средств биологического происхождения (т.н. биосимиляров или биодженериков). Для них исследования терапевтической эквивалентности – одно из условий регистрации. В скором времени такие препараты все чаще будут появляться на фармацевтическом рынке, поскольку сроки действия патентов на ряд оригинальных биопрепаратов (в т.ч. на низкомолекулярные гепараины) заканчиваются. В связи с этим некоторые дженериковые компании приступили к разработке производства биосимиляров, несмотря на то, что химическая структура и технология получения биосимиляров значительно сложнее, чем традиционных химических лекарственных средств. Поскольку биосимиляры имеют сложную трехмерную пространственную структуру, их количественное содержание в биологических жидкостях точно охарактеризовать достаточно трудно, поэтому принято считать, что для таких препаратов обычных исследований биоэквивалентности явно недостаточно. Это заставляет регуляторные органы требовать от производителей биосимиляров проведения как доклинических (токсикологических, фармакокинетических и фармакодинамических), так и клинических исследований (полного представления данных по эффективности и безопасности препарата), а также данных по изучению иммуногенности [9]. К препаратам биологического происхождения относятся гормоны, цитокины, факторы свертывания крови, моноклональные антитела, ферменты, вакцины и препараты, созданные на базе клеток и тканей и т.д.
«Дженериковая замена»
Необходимо отметить, что различия в лечебном эффекте оригинальных лекарств и дженериков или различных дженериков между собой, в принципе, допускаются рядом международных документов. Достаточно давно был введен термин «дженериковая замена», под которой понимают отпуск лекарственного препарата, коммерческое название которого отличается от выписанного врачом, а химический состав и дозировка действующего начала – идентична. В документах Всемирной Медицинской Ассамблеи делается предупреждение, что при отпуске препаратов, не полностью идентичных по химическому составу, биологическому действию или терапевтической эффективности пациент может столкнуться с неадекватным эффектом, т.е. с побочными реакциями или с недостаточной лечебной эффективностью. В этом документе обращается особое внимание на то, что государственные службы контроля должны информировать врачей о степени химической, биологической и терапевтической идентичности препаратов, выпускаемых одним или разными производителями, а службы контроля качества, существующие на предприятиях-производителях лекарств, обязаны следить за неуклонным соответствием выпускаемых препаратов стандартам химических и биологических свойств [10].
Возникает вопрос, почему, несмотря на установившиеся методы контроля дженериков, на рынок нередко попадают такие из них, которые явно не полностью соответствуют оригинальным препаратам ни по эффективности, ни по безопасности, а иногда и по тому и другому показателю [4]. Такая ситуация, к сожалению, достаточно типична для нашей страны [11, 12, 13, 14]. Окончательного ответа на этот вопрос пока нет, но, думается, главное заключается в нарушении тех самых принципов доклинической оценки дженериков, о которых было сказано выше. Хорошо известно, что в России стандарт GMP до сих пор не соблюдается при производстве большей части препаратов, выпускаемых в нашей стране (считается, что переход всех российских производителей лекарств на стандарт качества GMP должен произойти только к январю 2014 года), и уже одно это создает весомую причину для получения несоответствующих по качеству дженериков.
На что должен ориентироваться практический врач при выборе воспроизведенных препаратов
Возникает и более простой вопрос: что делать практическим врачам, выбирая лекарственный препарат, особенно в тех случаях, когда эта терапия является длительной и от качества которой может зависеть судьба больного, например, при вторичной профилактике сердечно-сосудиситых осложнений у кардиологических больных высокого риска. С одной стороны, все нормативные документы, а также экономическая целесообразность заставляют врача воспользоваться в первую очередь именно дженериком (если он зарегистрирован). С другой стороны, ряд четко спланированных клинических исследований (неконтролируемые исследования не в счет) свидетельствует о том, что далеко не все дженерики являются полноценными копиями. Этими фактами умело пользуются фармацевтические компании, утверждающие, что все дженерики – препараты неполноценные и, используя их, врач заведомо идет на назначение менее эффективной терапии [3].
Большинство российских специалистов, признавая изложенные выше факты, делают вывод о необходимости проведения прямых сравнительных исследований по изучению терапевтической эквивалентности с теми дженериками, которые уже зарегистрированы и чаще всего назначаются в клинике.Отделом профилактической фармакологии ФГУ ГНИЦ ПМ предпринята попытка создания реестра клинических контролируемых рандомизированных исследований, выполненных с дженериками в России [15].
Таким образом, с одной стороны, нет никаких оснований сомневаться в том, что создание дженерика – полной копии оригинального препарата – дело абсолютно возможное. Однако те или иные отклонения при разработке и производстве дженерика могут отразиться на его качестве. В идеале эти отклонения должны фиксироваться всей системой доклинического контроля, однако на практике, по-видимому, не всегда эта система четко соблюдается, что и приводит к появлению не полностью эквивалентных дженериков. В таких случаях единственным способом подтвердить качество дженерика является проведение методически грамотно спланированных сравнительных клинических испытаний по изучению терапевтической эквивалентности. Результаты таких исследований позволят также более точно ответить на вопрос о рациональности вмешательства как с точки зрения экономической эффективности, так и его доступности.
Источник


