Химико-токсикологический анализ лекарственных веществ в крови (плазме, сыворотке) методом высокоэффективной жидкостной хроматографии

библиографическое описание:
Химико-токсикологический анализ лекарственных веществ в крови (плазме, сыворотке) методом высокоэффективной жидкостной хроматографии / Крупина Н.А., Краснова Р.Р., Пашовкина Р.Н. // Мат. VI Всеросс. съезда судебных медиков. — М.-Тюмень, 2005.
код для вставки на форум:
Показана возможность применения метода высокоэффективной жидкостной хроматографии для скрининга и количественного определения лекарственных веществ в крови. Предложены условия изолирования лекарственных веществ из 0,5 мл крови.
Ключевые слова: высокоэффективная жидкостная хроматография, внутренний стандарт, жидкость жидкостная экстракция.
В настоящее время получило распространение злоупотребление медицинскими препаратами и использование лекарственных коктейлей в состав которых входит несколько лекарственных веществ из различных фармакологических групп. Доза каждого из принятых веществ может не достигать токсических концентраций, однако возрастает вероятность случайных и умышленных интоксикаций, нередко приводящих к смертельным исходам в результате фармакологического взаимодействия [1]. В связи с этим резко возрастает значение выбора метода изолирования и методики обнаружения при судебно – химическом исследовании на «неизвестное» вещество. Кроме того, необходимо правильно подобрать объект для исследования, его размерность, идентифицировать экстрагированное вещество с большой чувствительностью, специфичностью и провести количественное определение его. Количественное определение позволяет правильно интерпретировать полученные аналитические результаты и установить, имело ли место в данном случае отравление.
Современная высокоэффективная жидкостная хроматография — один из эффективных методов разделения сложных смесей на отдельные компоненты и проведения качественного и количественного анализа – компонентов разделяемой смеси. В конце 2003 года в лабораторию был приобретен высокоэффективный жидкостной хроматограф Agilent 1100 с диодно – матричным детектором (ДМД). При использовании ДМД проба сканируется каждые несколько миллисекунд, т.е. спектральная информация выдается практически постоянно. Диодная матрица постоянно регистрирует сигналы в ультрафиолетовой и видимой части спектра, обеспечивая таким образом, запись УФ – В — спектров в режиме сканирования. Это позволяет непрерывно снимать при высокой чувствительности неискаженные спектры быстро проходящих через спектральную ячейку компонентов. Данные, полученные от УФ – детектора одновременно на различных длинах волн, обрабатываются и оцениваются с помощью программного обеспечения, которое производит поиск в спектральной библиотеке, выделяет сигнал на определенной длине волны для повышения селективности, вычитает фон и осуществляет другие операции. Программное обеспечение может быть использовано для автоматической сверки УФ – спектров с известными, идентифицировать компоненты и проверять «чистоту» пиков путем сравнения спектров, записанных при начале и окончания выхода каждого пика. Такая автоматизированная система слежения идеальна для скрининга, поскольку позволяет отобрать для дальнейших исследований только «подозрительные» пики.
Целью нашего исследования был подбор оптимальных условий для изолирования лекарственных веществ из крови (плазмы, сыворотки), хроматографического скрининга лекарственных веществ и одновременного количественного определения их с использованием внутреннего стандарта.
Изучив зарубежную литературу по методам изолирования лекарственных веществ из биопроб с дальнейшим исследованием их методом ВЭЖХ [7], мы остановились на методе (несколько модифицировав его), который используется LNS Toxicologie (химико-токсикологическая лаборатория Люксембурга).
Нами также была создана база данных на лекарственные вещества, включающая в себя библиотеку УФ – спектров веществ, их абсолютные и относительные (относительно стандарта) [2 ]времена удерживания.
Экспериментальная часть
- Основа метода. Вещества из крови, сыворотки, плазмы экстрагируются с применением жидкость — жидкостной экстракции смесью н – гексана – дихлорметана – изо – пропанола при рН 9,5. Экстракты анализируются диодно – матричным детектором (ДМД) в градиентном режиме. Обнаружение веществ осуществляется по временам удерживания и УФ спектрам веществ. Количественное определение проводится с внутренним стандартом 5 – (4 – Метилфенил) – 5 – фенилгидантоином (MPPH).
- Объекты исследования. Объектами исследования являются кровь, сыворотка, плазма. Модельные исследования проводились на плазме, не содержащей лекарственных веществ, полученной из отделения переливания крови МОНИКИ им. М.Ф.Владимирского. В качестве реальных объектов использовали кровь, сыворотку живых лиц и кровь трупов, поступающих в отдел на исследование.
- Реактивы. Аммиачный буфер рН 9,5, насыщенный раствор сульфата аммония, н – гексан, дихлорметан, изо – пропанол, ацетонитрил для ВЭЖХ, МРРН, о-фосфорная кислота 85%, однозамещенный фосфат калия (Merck), стандартные растворы веществ (смесь): кофеин, фенобарбитал, димедрол, MPPH, феназепам в концентрации по 10мкг/мл.
- Приборы и принадлежности. Жидкостной хроматограф с диодно матричным детектором Agilent 1100, аппарат для встряхивания (горизонтальный тип встряхивания) 300 колеб./мин, центрифуга «Joan» 4000 об/мин, концентратор экстрактов с термоблоком, РН – тестер Checker, рН – метр (модификации рН 211), водоструйный насос, пипетки переменного объема 100 – 1000 мкл и 10 – 100 мкл с одноразовыми наконечниками, приспособление для фильтрования подвижной фазы.
- Условия обнаружения и определения. Исследование проводили на жидкостном хроматографе Agilent 1100 с диодно матричным детектором в градиентном режиме.
- Колонка:ZORBAX Eclipse XDB-C8 4,6 х 150 мм, 5 мкм;
- Предколонка ZORBAX SB – C18 4,6 х 12,5 мм, 5 мкм
- Температура термостата колонки 30 0 С
- Элюент А: вода деионизированная
- Элюент В: Ацетонитрил ВЭЖХ Градиент
- Элюент Д: Фосфатный буфер рН 3,8
- Градиент:
| Время (мин) | %В | %Д |
| 0 | 15 | 85 |
| 8 | 35 | 65 |
| 17 | 65 | 35 |
| 21 | 65 | 20 |
| 26 | 15 | 20 |
| 7 — 0 | 15 | 85 |
- Скорость потока: 1 мл / мин; от 7 – 0 минут кондиционирование.
- Длина волны: 220 нм. Объем вводимой пробы: 40 мкл.
Стандартные растворы. Стандартные растворы веществ и внутренний стандарт в концентрации 1мг/мл в метаноле, приготовленные весовым методом из стандартов веществ, чистота которых подтверждена методом хромато-масс спектрометрии.
Калибровочные растворы. Калибровочные образцы готовили из стандартных растворов (1мг/мл), с учетом терапевтических, токсических и летальных концентраций веществ в крови (С мг/л) [4]. Концентрации калибровочных растворов и линейность представлены в табл. 1.
Концентрация внутреннего стандарта (MPPH) 10 мкг/мл.
Концентрация калибровочных растворов и линейность.
| Вещество | С1 | С2 | С3 | С4 | С5 | Линейность (мг/л) |
| Амитриптилин | 0,2 | 0,5 | 1,0 | 2,0 | 5,0 | 0,2 — 5,0 |
| Диазепам | 0,1 | 0,5 | 1,0 | 5,0 | 0,1 — 5,0 | |
| Пропранолол | 0,05 | 0,5 | 2,0 | 5,0 | 0,05 — 5,0 | |
| Феназепам | 0,05 | 0,1 | 0,5 | 1,0 | 2,0 | 0,05 – 2,0 |
| Фенобарбитал | 5,0 | 10,0 | 20,0 | 50,0 | 100,0 | 5,0 – 100,0 |
Калибровка. Прибор с помощью программного обеспечения ChemStation автоматически проводит интегрирование пиков, при этом строится калибровочный график по данным калибровочной таблицы.
0,5 мл бланковой плазмы помещали в стеклянную пробирку на 12 мл, добавляли по 50 мкл (по 2 пробы) каждой концентрации калибровочных растворов веществ, тщательно перемешивали и затем настаивали в течение 5 минут для распределения в плазме. Добавляли 50 мкл МРРН (внутренний стандарт) в концентрации 10мкг/мл, тщательно перемешивали, 0,5 мл насыщенного раствора сульфата аммония, 0,5 мл аммиачного буфера (рН 9,5), Экстрагировали в течение 5 минут на аппарате для встряхивания (300кол/мин) с 5 мл смеси н – гексан – дихлорметан – изопропанол (60:40:2), центрифугировали 3 минуты при 4000 об/мин. Органическую фазу помещали в виалу на 4 мл и испаряли при температуре 40 0 С. Сухой остаток растворяли в 100 мкл подвижной фазы (15% ацетонитрила и 85% фосфатного буфера рН 3,8) и встряхивали в течение нескольких секунд на минишейкере IKA (2000/мин), 40мкл вводили в ВЭЖХ. Параллельно в вышеописанных условиях производили экстракцию и исследование бланковой плазмы.
Анализ исследуемого объекта. 0,5 мл исследуемой крови (сыворотки) помещали в стеклянную пробирку на 12 мл, добавляли 50 мкл внутреннего стандарта МРРН с концентрацией 10 мкг/мл, перемешивали. Далее анализировали аналогично описанному выше. Предварительно вводится стандартная смесь веществ: кофеин, фенобарбитал, димедрол, MPPH, феназепам в концентрации 10мкг/мл.
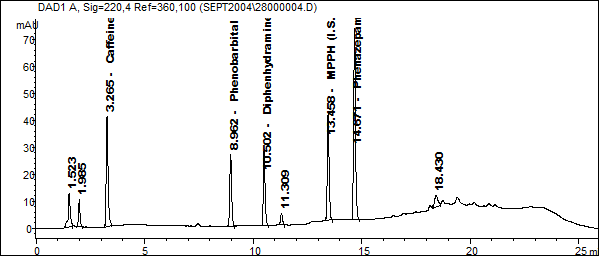
Рис.1. Стандартная смесь веществ (по 80 нг веществ во введённой пробе)
Идентификация лекарственных веществ осуществлялось по временам удерживания и библиотеке УФ – спектров.
Созданная база данных включает в себя следующие лекарственные вещества.
Амидопирин, амитриптилин, аминазин, анальгин, атенолол, атропин, верапамил, галоперидол, грандаксин, диазепам, дибазол, диклофенак, димедрол, карбамазепин, клозапин, кломипрамин, клофелин, кордиамин, кофеин, левомепромазин, левомицетин, миансерин, нитразепам, новокаин, нордазепам, но – шпа, оксазепам, папаверин, парацетамол, пропранолол, тиопентал, тиоридазин, трифтазин, феназепам, фенацетин, фенитоин, фенобарбитал, фтивазид, хлоразепат, хлордиазепоксид, циклобарбитал, циклодол, этаминал. Для некоторых из этих веществ были определены пределы обнаружения, определения,% выхода [6], и проведена статистическая обработка [3, 5].
Результаты
Предложенным методом выход лекарственных веществ основного характера в крови составил от 52,2 до 77%. (амитриптилин при С = 0,01 и 0,05мг/л выход 58 и 54,2% соответственно; димедрол при С = 0,025; 0,2; 10,0мг/л выход 77; 58,3; 70% соответственно; диазепам при С =0,1 и 1,0мг/л выход 54,1 и 53,2% соответственно; пропранолол при С = 0,05; 1,0; 5,0мг/л выход 57,1; 53,4; 70% соответственно; феназепам при С = 0,1; 0,5; 1,0мг/л выход 61,3; 57; 52,2% соответственно. Выход для фенобарбитала при С = 20,0 и 100,0 мг/л составил 16,5 и 15% соответственно. Пределы обнаружения и определения составили соответственно: 10нг/мл; и 50нг/мл (амитриптилин); 20нг/мл и 100 нг/мл (диазепам). 25нг/мл и 100нг/мл (димедрол); 10нг/мл и 50нг/мл (пропранолол); 30нг/мл и 80нг/мл (феназепам); 0,5мкг/мл и 2,0 мкг/мл (фенобарбитал)).
Статистическая обработка результатов (хi — индивидуальный результат, `Х – средняя арифметическая величина; S – стандартное отклонение; RSD (%) – относительное стандартное отклонение; Р – вероятность; Δ`Х – доверительный интервал) представлена в табл. 2.
Таблица 2 Статистическая обработка результатов.
| Наименование веществ | хi | Метрологические характеристики метода |
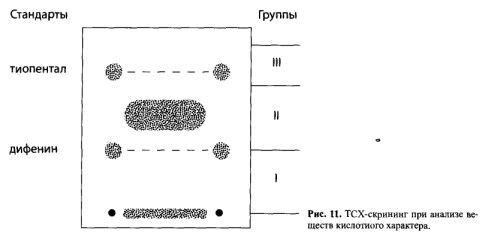
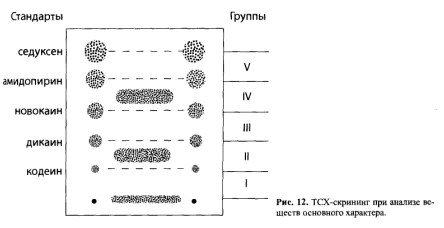
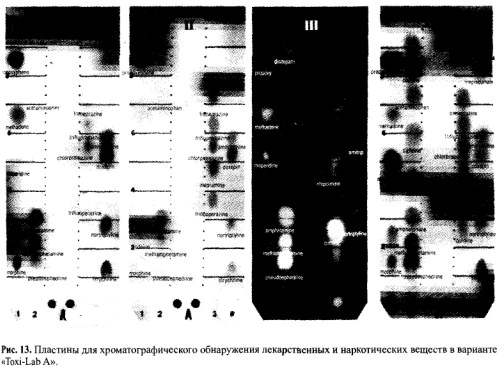
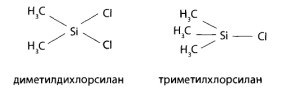
| Амитриптилин `Х =0,04 `Х =0,0473 Источник Глава 7. Методы обнаружения ядовитых веществ в извлечениях из объектов» data-shape=»round» data-use-links data-color-scheme=»normal» data-direction=»horizontal» data-services=»messenger,vkontakte,facebook,odnoklassniki,telegram,twitter,viber,whatsapp,moimir,lj,blogger»> Обнаружение ядовитых, сильнодействующих и наркотических веществ после их изолирования из биологических объектов является следующим этапом химикотоксикологического исследования. Несмотря на большое число реакций и методов качественного анализа, используемых в аналитической и фармацевтической химии, требуются некоторые особые подходы для их использования в токсикологической химии. Это объясняется особенностями объектов исследования, возможными примесями и требованиями к достоверности результатов. Химик-токсиколог для обнаружения в объекте какой-либо группы веществ или определенного вещества обязан использовать только те реакции и методы, которые апробированы на биологических объектах, рекомендованы для целей химико-токсикологического анализа с учетом всех факторов, влияющих на получаемые результаты. Только в этом случае полученные данные могут быть объективными и застрахованными от возможных ошибок, среди которых могут быть ошибки, связанные с получением ложноположительных или ложноотрицательных результатов. Ложноположительный результат – это заключение о наличии токсических, экзогенных веществ при анализе по определенной методике при фактическом их отсутствии. Это свидетельствует об использовании высокочувствительной, недостаточно специфичной методики и возможном отсутствии ее апробации на биологических объектах, особенно находящихся в стадии гнилостного разложения. Если какое-либо вещество или его метаболит присутствуют в объекте в концентрации ниже предела их обнаружения, то возможно получение ложноотрицательных результатов. Причиной этого чаще всего выступает фактор времени, т.е. интервал времени между контактом ядовитого вещества с организмом человека или животного и временем взятия и направления объекта на анализ. В этом случае даже правильно выбранная методика с высокой чувствительностью может дать отрицательный результат. Для обнаружения токсических соединений в биологических объектах в настоящее время применяются различные химические реакции, физические, физико-химические, биологические (фармакологические) и другие методы. Среди них выделяют методы и реакции предварительного анализа и методы и реакции подтверждающего исследования. Использование предварительного анализа или предварительных химических реакций преследует цель обнаружить или исключить из анализа группу веществ или какое-либо индивидуальное вещество. Таким реакциям или методам придают «судебно-химическое значение при отрицательном результате». Это означает, что если при использовании данного метода или реакции вещество или группа веществ не обнаружены, дальнейший анализ на эти соединения не проводят и в заключении указывают, что данное вещество или группа веществ в исследуемом объекте не найдены. Предварительные реакции и методы должны быть чувствительными, но необязательно специфичными. Среди них – скрининговые методы (ТСХ, Г’ЖХ – скрининг, им- мунохимические методы) и реакции, которые оцениваются как имеющие значение при отрицательном результате: групповые реакции осаждения, хромогенные реакции и др. Эти способы и реакции должны быть обязательно подтверждены химическими и физикохимическими методами. Предварительные реакции и методы, как правило, не позволяют определить индивидуальные вещества. Это требует проведения дополнительных исследований. В токсикологической химии такие исследования называют подтверждающим анализом, подтверждающими (частными) реакциями или частным внутригрупповым скринингом. Среди них методы ВЭЖХ, ГЖХ, ТСХ, УФ- и ИК-спектрометрия, спектрофотометрия в УФ области спектра, масс-спектрометрия, хроматомасс-спектрометрия, люминесцентный анализ, аналитические реакции разных типов (кислотно-основные, окислительно- восстановительные, комплексообразования), микрокристаллоскопический метод, фармакогностический анализ, фармакологические пробы, а также процессы осаждения, экстракции и др. Подтверждающие методы по чувствительности обычно выше или равны предварительным методам исследования. Это позволяет снизить возможность получения ложноотрицательных результатов. По специфичности подтверждающие методы обычно превосходят предварительные, что снижает количество ложноположительных результатов. При проведении химико-токсикологического анализа с использованием химического метода необходимо учитывать следующие обстоятельства. Любое чужеродное вещество в организме частично или полностью подвергается метаболизму, и поэтому его можно не обнаружить известными реакциями. В процессе изолирования ядовитые вещества извлекаются не полностью и могут быть частично потеряны, поэтому используемые реакции должны отличаться высокой чувствительно стью. Объекты, присылаемые на анализ, могут находиться в состоянии гнилостного разложения. Продукты разложения (птомаины) обычно извлекаются из объекта вместе с исследуемым веществом и могут искажать результаты анализа. Поэтому при анализе таких объектов использование химических реакций возможно после тщательной очистки извлечений. При комбинированных отравлениях (например, этиловым спиртом и его суррогатами) применение характерных реакций затруднено присутствием близких по химическим свойствам соединений. В этих случаях предусмотрено строгое соблюдение условий воспроизведения реакций (окисления, температурного режима и т.д.), чтобы предотвратить обнаружение одних соединений (более ядовитых) вместо других. Например, при несоблюдении условий проведения реакции окисления можно переоткрыть метиловый спирт за счет этилового спирта. Каждая используемая химическая реакция должна быть рекомендована в определенной модификации применительно к исследуемому объекту. Это особенно важно в тех случаях, когда проводят исследование объекта на «металлические» яды, поскольку многие из них являются микроэлементами и могут быть обнаружены в извлечениях. В таблице 5 даны пределы содержания токсикологически важных элементов в печени и почках – объектах, которые рекомендуются для анализа при подозрении на отравление «металлическими» ядами и содержат наибольшее количество микроэлементов. В связи с варьированием содержания элементов в тканях, для количественного определения металлов в минерализате для каждого катиона предложены не менее двух методов, которые позволяют определять их в широких пределах концентраций. По схеме дробного метода анализа предусматривается разбавление минерализата до концентраций, когда естественно содержащийся микроэлемент не может быть обнаружен, а концентрация попавшего в организм элемента останется достаточной для его обнаружения. При проведении реакций к минерализату добавляют определенный объем воды очищенной, а затем вносят соответствующие реактивы. Приведенные в таблице 6 разведения позволяют исключить обнаружение микроэлементов и одновременно снизить кислотность минерализата. Таблица 5. Пределы содержажания некоторых элементов в тканях организма человека (по данным А.Н. Крыловой) Границы естественного содержания, мг на 100 г органа Таблица 6. Разбавление минерализата при анализе его на «металлические» яды Объем минерализата, Объем воды Граница обнаружения, мг/100 г При анализе минерализата на «металлические» яды применяется избирательная экстракция в виде диэтилдитиокарбаматов с целью выделения меди, висмута, цинка и кадмия в органическую фазу, свободную от мешающих ионов. Специфичность экстракционных методик выделения этих катионов достигнута за счет использования правила ряда диэтилдитиокарбаматов металлов: Hg 2+ , Аg + , Cu 2+ , Ni 2+ , Со 2+ , Bi 3+ , Sb 3+ , Cd 2+ , Pb 2+ , Zn 2+ , Mn 2+ Согласно этому ряду каждый предыдущий металл вытесняет последующий из его соли с диэтилдитиокарбаминовой кислотой. Таблица 7. Маскировка некоторых мешающих ионов в химических реакциях на исследуемые металлы Со 2+ , Zn 2+ , Fe 3+ , Сd 2+ , Fe 2+ , Hg 2+ , Ag + Ag + , Pb 2+ , Fe 2+ , Bi 2+ , Fe 3+ , Sb 3+ , Cd 2+ , Hg 2+ Bi 3+ , Fe 3+ , Sb 3+ , Cd 2+ , Hg 2+ , Ag + и др. Селективная экстракция с дитизоном используется для обнаружения ионов свинца, серебра, таллия, цинка, которые при определенном значении pH среды образуют окрашенные дитизонаты. Для исключения влияния других ионов на результаты реакций при анализе минерализата используют прием маскировки мешающих ионов (табл. 7). С этой целью используют цианиды, фториды, фосфаты, тиосульфаты, тиомочевину, гидроксиламин, аскорбиновую кислоту, комплексон III (трилон Б) и др. Мешающие ионы образуют с этими веществами комплексные соединения и не влияют на результаты реакций, проводимых на исследуемые элементы. Абсолютно специфичных реакций очень мало. По правилам проведения химикотоксикологического анализа для заключения об обнаружении в извлечении из объекта ядовитого соединения необходимо провести не менее 3-4 характерных для данного вещества реакций. Например, чтобы сделать вывод об обнаружении в дистилляте этилового спирта, необходимо, чтобы три реакции дали положительный результат: образование йодоформа, окисление спирта до уксусного альдегида и образование эфира с уксусной кислотой (этилацетата). Результаты некоторых химических реакций оцениваются как «реакции Corpus delicti». Это реакции, продукты взаимодействия которых можно представить вместе с заключением об отравлении как неоспоримое доказательство обнаружения в объекте данного вещества. К числу таких реакций относятся: реакция образования берлинской лазури, результат обнаружения мышьяка в аппарате Марша, реакция образования «серебряного зеркала» и др. Полученный синий осадок берлинской лазури запаивают в небольшую пробирку или ампулу и представляют судебно-следственным органам для обоснованности заключения об обнаружении в объекте синильной кислоты. Восстановительная трубка аппарата Марша и микрофотографии налета на ней, имеющего характерную форму кристаллов в виде октаэдров, убедительное доказательство обнаружения мышьяка в исследуемом объекте. Для увеличения избирательности некоторых реакций и повышения их чувствительности иногда рекомендуется предварительное выделение ядовитого вещества из исследуемого раствора с использованием экстракции и реэкстракции. Например, чтобы отделить изоамиловый спирт от других «летучих» ядов и воды, его из дистиллята извлекают диэтиловым эфиром, эфир испаряют и в остатке обнаруживают изоамиловый спирт соответствующими реакциями. Фенол выделяют из дистиллята, подщелоченного гидрокарбонатом натрия, также путем экстракции диэтиловым эфиром, что позволяет повысить избирательность и чувствительность реакций на него. При химическом анализе на любое чужеродное соединение эксперт должен руководствоваться специально изданными и утвержденными методиками и соответствующими методическими указаниями. Комплексное использование различных методов предварительного и подтверждающего анализов позволяет надежно диагностировать причину отравления или болезненного состояния организма. Обнаружение ядовитых веществ и их метаболитов в процессе предварительного и подтверждающего химико-токсикологического анализа не дает однозначного ответа о количестве яда, попавшего в организм, времени его приема, фазах распределения. Результаты количественного определения найденного токсического соединения позволяют выбрать способ детоксикации и лечения пострадавшего при остром и хроническом отравлении. Выбор метода количественного определения ядовитого вещества в извлечении из объекта зависит: 7.1. Методы предварительного анализа 7.1.1. Понятие об аналитическом скрининге в химикотоксикологическом анализе Многообразие лекарственных, наркотических, ядовитых веществ, которые могут быть причиной отравлений, требует проведения предварительного отсеивающего исследования. В токсикологической химии такие исследования принято называть аналитическим скринингом. Скрининг (screening) в переводе с английского означает «просеивание», «отбор», «сортировка». Аналитический скрининг – это система методических приемов, позволяющих в ходе исследовательских операций исключить («отсеять») или определить группы веществ (индивидуальные соединения) на этапе предварительного исследования. Это позволяет построить дальнейший анализ в нужном направлении. Аналитический скрининг является эффективным при его соответствии определенным требованиям: Скрининг используется при анализе многокомпонентных смесей, а в случае ненаправленного анализа – при исследовании извлечения из биологического объекта на неизвестное токсическое вещество. В настоящее время понятие скрининга в токсикологической химии значительно шире. Скрининг – это поэтапное движение к выявлению индивидуального вещества путем последовательного исключения групп ядовитых веществ, а затем отсеивания веществ в обнаруженной группе до выявления конкретного соединения. Из современных скрининговых методов в практике химико-токсикологического анализа нашла широкое применение хроматография в тонких слоях сорбента (ТСХ) в нормально-фазовом и обращенно-фазовом вариантах. Этот метод доступен, прост в выполнении, отличается высокой чувствительностью, эффективностью, экспрессностью и достаточной специфичностью (избирательностью). ГЖХ-скрининг используется, в основном, при анализе летучих, лекарственных и наркотических веществ. Не потерял своего значения аналитический скрининг с использованием различных химических реакций. Имеются разработки по использованию в качестве скрининговых методов высокоэффективной жидкостной хроматографии (ВЭЖХ), абсорбционной спектроскопии, им- мунохимических методов, спектроскопии ядерного магнитного резонанса. При использовании этих методов необходима тщательная очистка извлечений от эндогенных соединений, способных исказить результаты анализа и засорить колонки хроматографов. 7.1.2. ТСХ-скрининг в нормально-фазовом варианте Метод используется в химико-токсикологическом анализе при исследовании веществ, изолируемых из объекта экстракцией и сорбцией (лекарственные, наркотические вещества, пестициды). Неподвижной фазой в ТСХ служит тонкий слой сорбента (0,1-0,5 мм), содержащий небольшое количество воды и нанесенный на пластинку (из стекла, фольги или полимера). Сорбентом чаще всего является силикагель или оксид алюминия, которые закрепляются на пластинке добавлением связующего компонента – гипса, крахмала и др. В качестве подвижной фазы (элюента, системы растворителей) предложены индивидуальные растворители или их смеси в определенных соотношениях. При движении элюента за счет капиллярных сил вверх по пластинке происходит разделение смеси веществ. Эффективность разделения зависит от сродства вещества к сорбенту и определяется коэффициентом распределения его между обеими фазами – подвижной и неподвижной. Чтобы обнаружить вещество на пластинке, используют различные способы детектирования: Для идентификации вещества используют величину Rf – отношение длины пробега анализируемого вещества к длине пробега растворителя (рис. 8). После хроматографирования измеряют расстояние от центра пятна до стартовой линии (отрезок АБ) и от линии фронта жидкости до стартовой точки (отрезок АВ). Отношение отрезков АБ к АВ обозначают величиной Rf: Величина Rf характерна для данного соединения на данном сорбенте в данной системе растворителей. Она зависит от качества и активности сорбента, толщины слоя, природы растворителей и их соотношения, а поэтому не всегда воспроизводима. Более надежной оценкой хроматографической подвижности вещества является величина R,. Для ее определения находят величину Rf исследуемого вещества и Rf вещества, принятого за стандарт. Их отношение малочувствительно к влиянию случайных отклонений в условиях эксперимента: Rs = Rf вещества / Rf стандарта Поэтому Rs — более воспроизводимая и относительно постоянная величина. Общий ТСХ-скрининг в нормально-фазовом варианте был разработан на кафедре токсикологической химии ММА им. И.М.Сеченова для систематического судебнохимического анализа биологических объектов на лекарственные и наркотические вещества, включенные по Приказу М3 СССР №1021 в обязательный круг исследования эксперта в случае ненаправленного анализа. Этот метод применим для веществ кислотного, нейтрального, слабоосновного и основного характера. ТСХ-скрининг предполагает разделение веществ в общих системах растворителей на хроматографические зоны с последующим исследованием каждой зоны, в которой были обнаружены те или иные соединения с использованием частных систем растворителей. Как мы уже отмечали ранее, лекарственные и наркотические вещества из биологических объектов изолируются путем настаивания с подкисленным спиртом (методы Стаса-Отто, Саломатина), нейтральным ацетоном (метод Карташова), подкисленной водой (методы Васильевой, Степанова-Швайковой, Поповой, Крамаренко), подщелоченной водой (метод Валова). Из полученных извлечений (вытяжек) вещества кислотного, нейтрального и слабоосновного характера экстрагируются диэтиловым эфиром или хлороформом при рН=2-2,5, а вещества основного характера – при рН=8-10. Хлороформные (эфирные) экстракты упаривают до небольшого объема и исследуют. Применительно к веществам, включенным в учебную программу по токсикологической химии, схему ТСХ-скрининга в общих системах растворителей можно представить следующим образом (по Б.Н.Изотову). ТСХ-скрининг веществ кислотного, нейтрального и слабоосновного характера Цель: выявить наличие токсикологически важных лекарственных веществ в извлечениях и определить их принадлежность к определенной группе соединений. Условия анализа: сорбент – закрепленный слой силикагеля. Система растворителей: хлороформ ацетон (9:1). Время насыщения камеры парами системы – 15-20 мин. Длина пробега системы растворителей по пластинке 10 см. На стартовую линию хроматографической пластинки наносят (см. рис. 9): Реагенты-проявители для обнаружения веществ на пластинке: – слой сорбента полос А, Б и В обрабатывают 5% раствором сульфата ртути (II) и 0,1% раствором дифенилкарбазона в хлороформе. Производные барбитуровой кислоты проявляются в виде фиолетовых пятен. Пластинку нагревают горячим феном ( 50°С). Пятна обесцвечиваются. Все вещества кислотного, нейтрального и слабоосновного характера делятся на 3 хроматографические зоны в зависимости от значения величины Rf в указанной общей системе растворителей. Зона 1 включает 2 вещества, имеющие значение Rf 0-0,25. В этой зоне обнаруживаются антипирин (Rf 0,19) и кофеин (Rf 0,25). Зона 2 включает 7 веществ, имеющих значение Rf 0,31-0,41. В этой зоне обнаруживаются фенобарбитал (Rf 0,31), барбитал (Rf 0,32), нитразепам (Rf 0,35), барбамил (Rf 0,37), этаминал-натрий (Rf 0,37), бутобарбитал (Rf 0,41), циклобарбитал (Rf 0,41). Зона 3 включает вещества, имеющие значение Rf 0,41-0,64. В этой зоне обнаруживается диазепам (Rf 0,62). Полоса Г на пластинке используется для соскабливания сорбента (заштрихованная часть), элюирования веществ подходящим растворителем (1 зона – метанолом; 2 и 3 зоны – ацетоном) и последующего хроматографического исследования в частных системах растворителей, в которых все вещества обнаруженной группы соединений хорошо разделяются между собой. ТСХ-скрининг в анализе веществ основного характера Цель: выявить наличие в извлечении из объекта веществ, являющихся соединениями основного характера. Условия анализа: сорбент – закрепленный слой силикагеля. Система растворителей: хлороформ – диоксан – ацетон – 25% раствор аммиака (45:47,5:5:2,5), время насыщения камеры парами системы – 15 мин. Пробег растворителя по пластинке – 10 см. На стартовую линию хроматографической пластинки наносят (рис. 10): – полоса А – растворы стандартов (например, атропина и новокаина); – полоса Б – 1/25 часть хлороформного экстракта, полученного из водного извлечения при рН=8-10; Реагенты-проявители: слой сорбента полос А, Б, В (при закрытой полосе Г) последовательно обрабатывают: – 10% раствором хлорида железа (III). Возможно обнаружение производных фенотиазина и пиразола; – 5% раствором нитрита натрия, содержащим 3% хлорной кислоты. Обнаруживают ярко выраженные окрашенные пятна производных фенотиазина; – 10% раствором серной кислоты и просматривают в УФ-лучах. Возможно обнаружение хинина по голубой флуоресценции; – модифицированным реактивом Драгендорфа. Вещества основного характера проявляются в виде оранжевых пятен на бледно-желтом фоне. Соединения основного характера делятся на пластинке на 4 хроматографические зоны, в зависимости от значения Rf. Зона 1 включает 8 веществ со значением Rf 0,12-0,36. В этой зоне обнаруживаются пахикарпин (Rf 0,12), морфин (Rf 0,14), атропин (Rf 0,15), эфедрин (Rf 0,18), хинин (Rf 0,25), стрихнин (R 0,25), кодеин (Rf 0,28), этилморфин (Rf 0,31). Зона 2 включает 3 вещества со значением Rf 0,5-0,58. В этой зоне обнаруживаются этаперазин (Rf 0,51), антипирин (Rf 0,54), кофеин (Rf 0,56). Зона 3 включает 7 веществ со значением R, 0,63-0,83. В этой зоне обнаруживаются хлордиазепоксид (Rf 0,63), дипразин (Rf 0,66), новокаин (Rf 0,7), тиоридазин (Rf 0,72), аминазин (Rf 0,75), промедол (Rf0,76), нитразепам (R, 0,77). Зона 4 включает 4 вещества со значением Rf 0,87-0,98. В этой зоне обнаруживаются левомепромазин (Rf 0,87), папаверин (Rf 0,91), диазепам (Rf 0,95), кокаин (Rf 0,98). Полосу Г (заштрихованная часть) соскабливают, элюируют соответствующим растворителем, для хроматографической зоны 1 применяют метанол – диэтиламин (9:1), для 2, 3,4 хроматографических зон метанол – 25% раствор аммиака (9:1). Затем проводят исследование полученных элюатов в частных системах растворителей, в которых вещества данной зоны хорошо разделяются. Если ни в одной из зон ни с одним из реактивов пятна на пластинках не обнаружены, делают заключение о ненахождении в объекте исследуемых веществ. При обнаружении пятна в какой-либо из зон проводят основное исследование с использованием подтверждающих реакций и методов. При экспресс-анализе острых отравлений наркотическими и психотропными веществами ТСХ-скрининг проводят на двух пластинках в двух системах растворителей: На обе пластинки «Силуфол УФ-254» наносят аликвоты извлечений из объекта и стандартные растворы морфина, кокаина, кодеина, аминазина, димедрола, амитрип- тилина и помещают в указанные системы растворителей. Когда системы растворителей достигают высоты 10 см, пластинки вынимают и, после высушивания, просматривают в УФ-свете. Отмечают пятна стандартных веществ и на том же уровне пятна, полученные из вытяжки (извлечения из объекта). Дополнительно на эти зоны наносят капельно реактивы на предполагаемые вещества. Например: Хроматографирование в двух системах растворителей разной полярности позволяет достаточно надежно провести групповое, а в ряде случаев индивидуальное обнаружение наркотических и психотропных веществ. ТСХ-скрининг токсических веществ кислотного и основного характера по методу В.А.Карташова Исследование проводят на специально приготовленных хроматографических пластинках по следующей методике. К 3 г силикагеля марки КСК добавляют 0,2 г гипса и 7,5 мл воды для веществ кислотного характера или 0,025 М раствора гидроксида калия для веществ основного характера. Смесь перемешивают в ступке до однородной массы, наносят на обезжиренную стеклянную пластинку размером 9х 12 см, подсушивают и активируют при 120°С 30 мин. Вещества кислотного характера Сухой остаток, полученный при изолировании по методу В.А.Карташова и содержащий вещества кислотного характера, растворяют в небольшом объеме хлороформа и количественно наносят на стартовую линию хроматографической пластинки (рис. 11). По краям стартовой линии наносят хлороформные растворы «стандартов» (дифенин и тиопентал). Условия анализа. Система растворителей: ацетон – н-гексан – диэтиламин (10:10:1). После высушивания пластинку обрабатывают раствором сульфата ртути. Вещества кислотного характера проявляются в виде полосы, а «стандарты» – в виде пятен белого цвета. Таблица 8. Хроматографические группы веществ кислотного характера Таблица 9. Распределение веществ по хроматографическим группам 1 группа 22 вещества 2 группа 16 веществ 3 группа 7 веществ 4 группа 18 веществ 5 группа 12 веществ 6 группа 5 веществ Анабазин Аминазин Дикаин Скополамин и др. Феназон Папаверин Эфедрин Седуксен Резерпин и др. По полученным данным вещество относят к одной из трех хроматографических групп (табл. 8). Зону силикагеля, содержащую исследуемое вещество, соскабливают, элюируют хлороформом, который далее исследуют с помощью частных реакций и физико-химических методов. Вещества основного характера К экстракту из щелочного раствора, полученного при изолировании по методу В.А.Карташова, добавляют 2 капли 10% спиртового раствора хлороводородной кислоты и выпаривают при 40°С досуха. Остаток растворяют в небольшом объеме хлороформа, насыщенного аммиаком, и наносят в виде полосы на стартовую линию хроматографической пластинки (рис. 12). Справа и слева в две точки на линию старта наносят смесь из 5 «стандартов»: кодеина, дикаина, новокаина, амидопирина и седуксена. Условия анализа. Подвижная фаза – ацетон. После высушивания пластинку рассматривают в УФ-лучах (светофильтры 254 и 360 нм), отмечают флуоресцирующие зоны, затем обрабатывают реактивом Драгендорфа. «Стандарты» образуют на пластинке пятна оранжевого цвета, и пластинка делится на 6 зон. Исследуемое вещество, проявляющееся; на пластинке в виде окрашенной полосы, попадает в одну из шести хроматографических групп (см. табл. 9). Если ни в одной из зон не обнаруживаются полосы оранжевого цвета, делают заключение, что в исследуемом объекте вещества основного характера отсутствуют. Если обнаружено пятно, окрашенную зону снимают с пластинки, добавляют гидроксид натрия и экстрагируют диэтиловым эфиром. Эфир испаряют и проводят основное исследование на вещества, относящиеся к определенной группе, используя частные химические реакции и различные физико-химические методы. 7.1.3. ТСХ-скрининг в варианте «Toxi-Lab АВ» В этом варианте модифицирован классический метод тонкослойной хроматографии. Это специально разработанная аналитическая система, которая предназначена для исследования одной пробы объекта (мочи) на наличие наркотических средств, психотропных и сильнодействующих веществ. Эта система обеспечивает стадию пробоподготовки, экстракцию, концентрирование, разделение веществ и их обнаружение. Эта система представлена тремя наборами экстракционных пробирок с органическим растворителем определенного состава, набором ТСХ-пластин, концентрационных дисков и сосудами для обнаружения веществ: Методика использования системы включает несколько стадий: На рисунке 13 представлены пластины из каталога системы «Toxi-Lab А» для веществ основного и нейтрального характера. Пластины I, II и III детектированы с помощью реактивов Марки-Манделина. Сначала пластинки выдерживали в парах 37% формальдегида и затем погружали в концентрированную серную кислоту, содержащую 0,1% ванадата ммония. Пластина III промыта погружением в воду и освещена УФ-лучами при 254 нм, IV пластина обработана модифицированным реактивом Драгендорфа. На пластины в места 1, 2, 3, 4 фирмой-производителем нанесены растворы стандартных соединений лекарственных и наркотических веществ. Идентификация веществ на пластинах проводится по окрашенным в определенные цвета или флуоресцирующим пятнам путем сравнения с характеристиками стандартных соединений, нанесенных на пластины производителем. Метод ТСХ в варианте «Toxi-Lab» позволяет не использовать один из факторов плохой воспроизводимости результатов ТСХ-анализа – нанесение пробы на пластинку в ее классическом варианте. Многостадийное детектирование исключает возможность неправильной интерпретации получаемых результатов. Предел обнаружения веществ составляет 1 мкг/мл (морфин). В наборе реактивов и оборудования в данной системе предусмотрено наличие каталога с образцами результатов анализа более чем для 100 различных токсических веществ и их метаболитов. 7.1.4. ТСХ-скрининг в обращенно-фазовом варианте (ОФТСХ) Метод разработан сотрудниками кафедры токсикологической химии Пермской государственной фармацевтической академии. Сорбентом является тонкий слой силикагеля «Сорбтон-2» с привитой алкильной фазой С2 и «Плазмохром» с привитой фазой С.. Привитые фазы придают сорбенту гидрофобные свойства. В качестве связующего компонента использована соль кремниевой кислоты, за счет которой слой оказался достаточно прочным и позволил применять для детектирования последовательно несколько реагентов с целью обнаружения различных групп химических соединений. Для веществ кислотного, нейтрального и слабоосновного характера авторами предложена система растворителей этанол – вода (3:7), для веществ основного характера – этанол – вода – 25% раствор аммиака (6:5,5:0,5). Для обнаружения токсических веществ на хроматографических пластинках в качестве реактивов рекомендуются: Предел обнаружения веществ на пластинках при обращенно-фазовом варианте в 2-3 раза меньше по сравнению с нормально-фазовым вариантом ТСХ. Идентифицируют вещества на пластинках по величине Rf или по величине R, при хроматографировании в присутствии вещества-«стандарта». 7.1.5. Внутригрупповой ТСХ-скрининг в частных системах растворителей Любой вариант ТСХ может быть использован для определения индивидуальных веществ в извлечениях из биологического материала. Для этого необходимо использовать условия, разработанные для узкого круга ядовитых веществ. В качестве «стандартов» берут вещества из этой же группы. Как пример приводим определение индивидуального барбитурата, если на предварительном этапе выявлено наличие барбитуратов в извлечении (методика описана Б.Н.Изотовым). На пластинку со слоем силикагеля, как показано на рисунке 14, наносят растворы стандартов: барбитала, фенобарбитала, барбамила, этаминала, бутобарбитала (по 2 капли 1 % растворов), а рядом – извлечение из объекта или часть элюата, полученного с пластинки (полоса Г на рис. 10). Для обнаружения места расположения пятен используют специальные реактивы, которыми опрыскивают с помощью пульверизатора пластинку после ее проявления в системе растворителей и высушивания. Система растворителей (частная для барбитуратов): хлороформ – н-бутанол – 25% раствор аммиака (70:40:5). Сорбент – силикагель КСК, забуференный 0,1 М раствором борной кислоты. Для производных барбитуровой кислоты пластинку обрабатывают в начале раствором сульфата ртути (II), а затем раствором дифенилкарбазона в хлороформе. Барбитураты проявляются в виде сине-фиолетовых или красно-фиолетовых пятен и имеют следующие значения Rf: барбитал – 0,55; фенобарбитал – 0,40; барбамил – 0,90; этаминал – 0,95; бутобарбитал — 0,65. Пятно стандарта и пятно анализируемого вещества должны иметь одинаковую окраску и одинаковое значение Rf. Можно привести такие же примеры по определению индивидуальных веществ из других трупп токсикологически важных веществ (морфинана, хинолина, фенилалки- ламина и др.). 7.1.6. Газожидкостная хроматография Газожидкостная хроматография является одним из эффективных методов при проведении скрининга. Основным условием использования этого метода является летучесть соединений при температуре испарителя. Во многих методиках рекомендуется переводить исследуемые вещества в легколетучие соединения путем их дериватизации. В таких случаях метод ГЖХ является универсальным способом для проведения скрининга. Газожидкостная хроматография – это один из видов распределительной хроматографии. Разделение веществ происходит в специальных колонках, заполненных твердым носителем, представляющим собой пористый материал природного или синтетического происхождения (пемза, кизельгур, полисорб, целит и др.). Зернистый носитель набивается в колонку – трубку малого диаметра длиной от нескольких сантиметров до нескольких метров. Колонки изготовляют из нержавеющей стали, меди, алюминия, стекла и других материалов. Твердый носитель имеет тонкий слой неподвижной фазы. Неподвижная жидкая фаза может быть неполярной – апиезоны (L, М, N, SE-30, OV-1, OV-101). Это углеводороды и полимеры, в основном, диметил- и триметилсилана: Неподвижная фаза может быть полярной и чаще всего представлена полиэтиленгли- колем с массой 20 ООО – карбовакс-20М, фенилметилсиланом OV-17, цианоэтилсиланом ХЕ-60 и др. Подвижной фазой является инертный газ, чаще всего азот или гелий. Принцип устройства газожидкостного хроматографа приведен на рисунке 15. Исследуемый объект вносят в дозатор (3) в количестве нескольких микролитров с помощью микрошприца. Вещества переносятся газом-носителем (7) в колонку (5). На колонке происходит разделение компонентов смеси. Разделение смеси зависит от величины коэффициентов распределения веществ между подвижной и неподвижной фазами и от эффективности колонки. Эффективность колонки выражается числом теоретических тарелок. Под «теоретическими тарелками» понимают количество теоретических ступеней, на которых устанавливается равновесие между подвижной и неподвижной фазами. Чем больше таких ступеней на единицу длины колонки, тем лучше происходит разделение веществ. Из колонки разделенные вещества поступают в детектор (6). Детектор – часть прибора, которая регистрирует интенсивность определенных свойств бинарных смесей (компонент + газ-носитель). Изменение интенсивности сигнала детектора свидетельствует об изменении состава газовой смеси. Детекторы могут быть настроены на измерение интенсивности различных физико-химических свойств системы (плотности, теплопроводности, теплоты сгорания, ионизации и т.д.), которые преобразуются в электрический сигнал детектора, регистрируемый самописцем (7). В практике химико-токсикологического анализа используют катарометр – детектор теплопроводности. Катарометром измеряют разность теплопроводностей чистого газа- носителя и смеси газа-носителя с анализируемым веществом. Чувствительность определения токсических веществ с помощью катарометра находится в пределах 10 -3 — 10 -5 г. Пламенно-ионизационный детектор (ПИД). Принцип действия основан на измерении тока, возникающего при ионизации молекул органических веществ в пламени водорода. При горении чистого водорода ионы почти не образуются, поэтому электропроводность водородного пламени очень низка. Органические вещества, сгорающие в пламени водорода, образуют ионы или радикалы. Появление заряженных частиц обусловливает электропроводность пламени. Увеличение электропроводности повышает силу ионного тока, которая отображается на хроматограмме в виде пика. Чувствительность ПИД — 10 -9 – 10 -12 г. Хроматограмму двух растворенных веществ можно представить следующим образом (рис. 16). На кривой разделения каждому пику свойственны следующие параметры. Высота пика (2) – это расстояние от вершины пика до его основания (3). Площадь пит (4) – площадь, заключенная между контуром пика и его основанием. Основание пика (3) – отрезок нулевой линии (1) между крайними точками пика. Качественной характеристикой в газожидкостной хроматографии считаются: В основе идентификации веществ с помощью ГЖХ – сравнение индекса удерживания неизвестного вещества с индексом удерживания известного соединения. 7.1.6.1. ГЖХ-скрининг в анализе лекарственных и наркотических веществ в извлечениях из мочи Исследуемые вещества выделяют из мочи путем экстракции или сорбции. Для ГЖХ- скршшнга достаточно использовать часть извлечения из объекта, которая соответствует по объему 10 мл мочи (представлено по методикам, описанным С.К.Ереминым). Условия хроматографического анализа: прибор ЛMХ-80, детектор термоаэрозольный (ТАД), термоионный или беспламенный азотно-фосфорный (NPD), колонка стеклянная, силанизированная, длиной 1 м, внутренний диаметр – 2-3 мм, сорбент – 3% SE-30 на хромосорбе W (HP) – 80-100 меш., скорость газа-носителя (азота) для ТАД 45 мл/мин и для NPD – 40 мл/мин (гелия), эффективность колонки 1200 т.т. (ТАД) и 1300 (NPD), температура детектора – 300°С, температура испарителя 250°С, температура испарителя по линейной программе от 130 до 290°С, объем пробы – 2,5 мкл. Для идентификации хроматографических пиков определяют индекс удерживания (Ix). С этой целью используют в качестве стандартов смесь н-алканов СnН2n+2 с числом атомов углерода от 10 до 32, которым присвоены значения индексов, равные 100n (n – число атомов углерода в алкане). Расчет проводят по формуле: Ix = 100n + (In+1 – In) · [(lg tx – lg tn) / (lg tn+3 – lg tn)] Вместо исправленного времени удерживания иногда используют в расчетах истинные удерживаемые объемы. При работе с другими детекторами вместо н-алканов используют в качестве стандартов смесь азотсодержащих веществ, индексы удерживания которых также предварительно определены по н-алканам при тех же условиях (см. табл. 10). Стандартное отклонение измеренных индексов удерживания находится в пределах 15-20 единиц. При скрининге «поисковое окно» должно находиться в пределах ±50-60 единиц I, если сравнивается I неизвестного соединения с I известных веществ. Таблица 10. Смесь анализируемых веществ для расчета индексов удерживания Вещество Индекс удерживании по н-алканам Источник  От скованности в шее можно легко избавиться.  Язвенник обыкновенный Язвенник обыкновенный – это двулетнее  1. Лечение зубов Нет людей, которые не боятся стоматолога. |