- Химическая разработка лекарственных препаратов
- Создание лекарств
- Содержание
- Сырье и действующее вещество [ править | править код ]
- Цели выделения очищенных веществ [ править | править код ]
- Растения как источник лекарств [ править | править код ]
- Разработка фармакологических препаратов [ править | править код ]
- Этапы разработки лекарственных препаратов
- Этапы разработки лекарственных средств [ править | править код ]
Химическая разработка лекарственных препаратов
Разработка лекарственного средства начинается с синтеза новых химических соединений. Вещества со сложной структурой можно получить из различных иа очников, например растений (сердечные гликозиды), тканей животных (гепарин), микробных культур (бензилпенициллин), культур человеческих клеток (урокиназа), или посредством генно-инженерных технологий (человеческий инсулин).
Чем больше ясности во взаимоотношениях структуры и активности, тем более направленным оказывается поиск новых веществ.
а) Доклинические испытания дают информацию о биологических эффектах новых веществ. Начальный скрининг может включать биохимические и фармакологические исследования (анализ связывания с рецептором) или эксперименты на культурах клеток, изолированных клетках и изолированных органах.
Поскольку эти модели не способны воспроизвести сложные биологические процессы, происходящие в интактных организмах, любое потенциальное лекарственное средство необходимо проверить на животных. Только эксперименты на животных позволяют выяснить, возникаютли желаемые эффекты при дозах, не вызывающих токсичности или сопровождающихся слабой токсичностью. Цель токсикологических исследований заключается в том, чтобы оценить:
1) токсичность, обусловленную кратковременным или длительным приемом;
2) генетические повреждения (генотоксичность, мутагенез);
3) развитие опухолей (канцерогенность);
4) возникновение врожденных дефектов (тератогенность).
В экспериментах на животных также оценивают всасывание, распределение, метаболизм и элиминацию (фармакокинетика) изучаемых веществ. На уровне доклинического изучение лишь у малой части новых веществ обнаруживается потенциал для применения у человека.
Фармацевтическиетехнологии предлагают методы изготовления лекарственных форм.
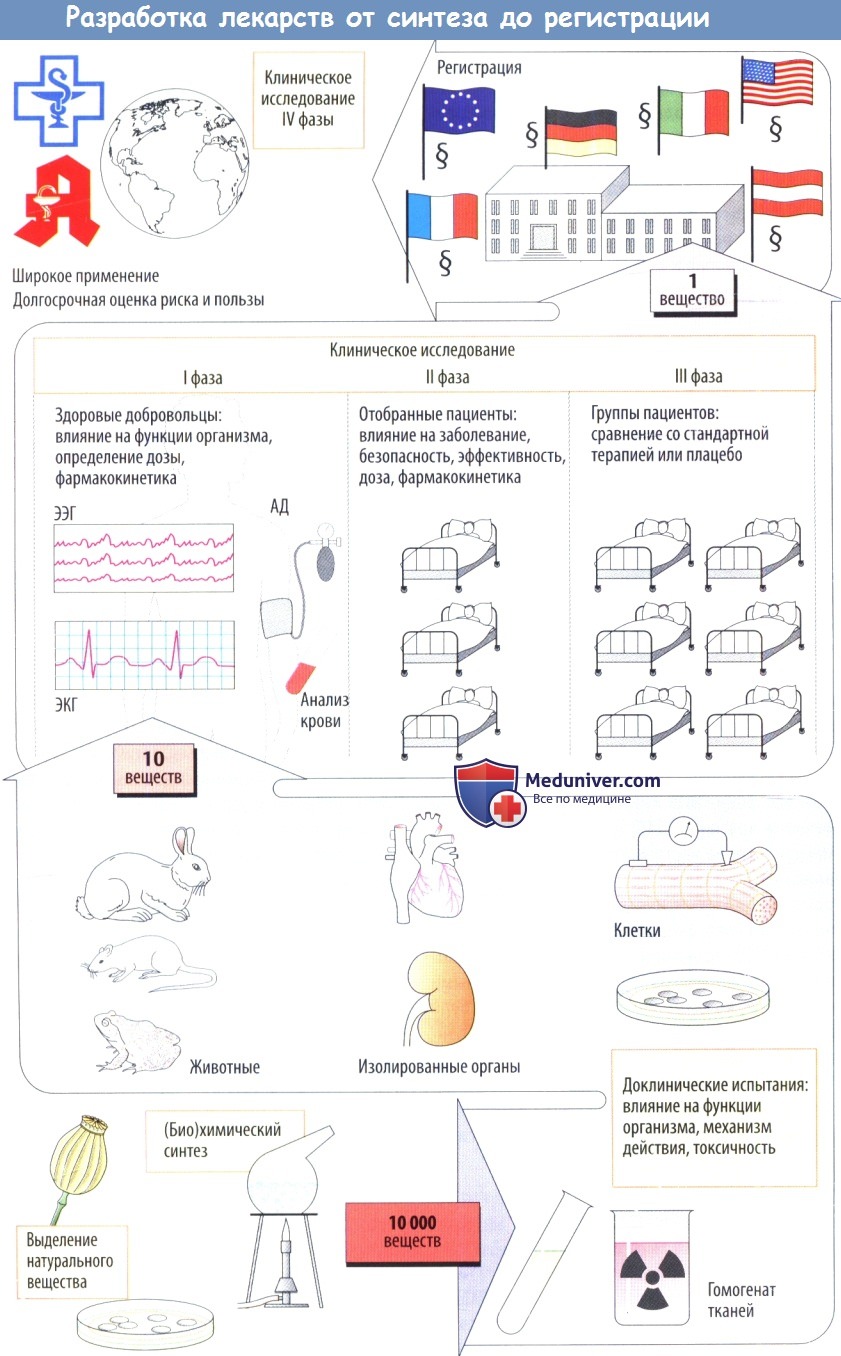
б) Клинические испытания начинаются с исследований I фазы, в которых участвуют здоровые лица; цель этих исследований — определить, будут ли эффекты, наблюдаемые у животных, также возникать у людей. Кроме того, на данном этапе определяется дозозависимость клинических эффектов.
Во II фазе потенциальные лекарственные средства сначала проверяют на отобранных пациентах на терапевтическую эффективность при заболевании, для лечения которого эти препараты предназначались. Если полезное действие очевидно, а частота побочных эффектов приемлема, начинается III фаза, в которой участвует более крупная группа пациентов, у которых новое средство сравнивают с традиционными методами лечения сточки зрения терапевтического результата.
Как форма экспериментов на людях, эти клинические исследования подвергаются анализу и одобрению этическими комитетами медицинских учреждений в соответствии с международными правилами проведения (Хельсинкской, Токийской и Венецианской декларациями). Во время клинических исследований выясняется, что многие вещества нельзя использовать. Как правило, в конце концов примерно из 10 000 вновь синтезированных веществ остается только одно.
в) Решение зарегистрировать новое лекарственное средство выносится национальным регуляторным органом (Food and Drug Administration в США, Health Protection Branch Drugs Directorate в Канаде, комиссией ЕС вместе с European Medicines Agency в Великобритании), которому производители должны подавать регистрационные документы.
Заявители должны документально подтвердить результатами соответствующих испытаний (доклинических и клинических), что критерии эффективности и безопасности удовлетворены и что лекарственные формы продукта (таблетки, капсулы и т. д.) соответствуют всем стандартам контроля качества.
После регистрации новое лекарственное средство может продаваться под торговым названием, оно должно быть доступным, выписываться врачами и отпускаться фармацевтами. В это время наблюдение продолжается в форме постмаркетинговых исследований (IV фаза клинических исследований)
г) Фармакологический надзор — действия, направленные на то, чтобы выявлять и устранять связанные с препаратом риски во время проведения клинических исследований и последующего его выхода на рынок. Фармаконадзор включает отчеты о предполагаемых случаях нежелательных реакций, направляемые в национальные регуляторные органы.
На основе длительного опыта применения можно правильно оценить соотношение риска и пользы и, следовательно, терапевтическую ценность нового лекарственного средства. Если новый препарат имеет небольшое преимущество перед существующими, необходимо принимать во внимание соотношение затрат и пользы от применения лекарственного средства.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Источник
Создание лекарств
Содержание
Сырье и действующее вещество [ править | править код ]
Вплоть до конца XIX в. при лечении болезней применяли лекарства, которые готовили из минерального или природного сырья, чаще всего из высушенных растений или их частей, но нередко в дело шли и свежие растения. Они могли содержать не только вещества, оказывающие целебное (терапевтическое) действие, но и вещества с отравляющим (токсическим) эффектом.
Для получения растительного сырья, которое могло бы храниться и использоваться для изготовления лекарства на протяжении всего года, растения высушивали или заливали растительным маслом либо спиртом. При высушивании растения или продукта растительного или животного происхождения получается лекарственное сырье.
При помещении растений или их частей в спирт (этанол) получается настойка (тинктура): под воздействием спирта происходит вытяжка (экстракция) фармакологически активных веществ из растения. Настойки не содержат всего набора представленных в растении веществ, а лишь те из них, которые растворяются в спирте.
При назначении препарата природного происхождения или экстракта пациент принимает сразу несколько веществ, которые оказывают различное действие. При этом содержание действующих веществ и их соотношение в природном сырье могут сильно варьировать в зависимости от происхождения растения (места произрастания), времени сбора, а также длительности и условий хранения. По этим же причинам соотношение различных веществ в лекарственном препарате может также значительно варьировать.
В фармакологических лабораториях из различных природных продуктов были выделены многие фармакологически активные вещества в виде химических соединений.
Цели выделения очищенных веществ [ править | править код ]
- Идентификация одного или нескольких активных веществ.
- Изучение биологического действия (фармакодинамика) отдельных веществ; анализ их «судьбы» в организме (фармакокинетика).
- Подбор точной и постоянной дозы вещества в случае его использования для лечения.
- Химический синтез позволяет получать лекарства вне зависимости от поставок природного сырья. На синтетических препаратах изучаются связи между химическим строением вещества и его действием, что в конце концов и приводит к открытию новых соединений с интересными фармакологическими свойствами.
Благодаря изменению химической структуры синтезированные вещества часто обладают более сильным фармакологическим действием.
Растения как источник лекарств [ править | править код ]
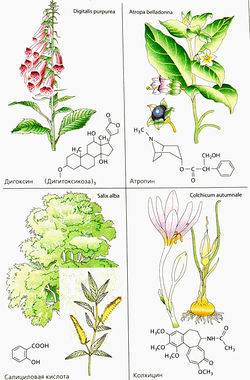
Еще в древности человек пытался лечить болезни и раны, используя растения. Некоторые рецепты дошли до нас из античных времен. В средневековых «травниках» в качестве лечебных средств также предлагались всевозможные растения. В современной медицине, где от каждого лекарства ожидают определенного действия, из сотен лекарственных растений востребованы только немногие. Рассмотрим четыре лекарственных растения, которые применялись для лечения еще в «донаучное» время, причем вещества, выделенные из этих растений, и сегодня входят во многие лекарства как важный активный компонент.
- В средние века против «водянки» использовали такие повсеместно распространенные растения, как наперстянка пурпурная (Digitalis purpurea), ландыш майский (Convallaria majalis), морозник черный
- Произрастающая в Центральной Европе красавка (Atropa beladonna, семейство пасленовых) содержит алкалоид атропин и в небольших количествах скополамин. Действие экстракта этого растения было известно еще в античности; женщины использовали его как косметическое средство (при закапывании в глаза возникал эффект расширения зрачков). В XIX в. алкалоиды были выделены, расшифрована их химическая структура и установлен механизм действия этих веществ. Сегодня атропин применяют как основной антагонист ацетилхолинового рецептора мускаринового типа.
- Кора ивы белой (Salix alba) содержит производное салициловой кислоты. Настойки этого растения готовили еще в древности. Салициловая кислота как действующее вещество этой настойки, применяемой в народной медицине, была выделена в XIX в. В современной медицине эта кислота все еще используется для наружного применения (кератолитическое действие), но не перорально как болеутоляющее, жаропонижающее или противовоспалительное средство. После получения ацетилсалициловой кислоты (аспирина) путем ацетилирования салициловой кислоты (около 1900 г.) это лекарственное средство стало активно применяться для перорального приема.
- Осенний безвременник (Colchicum autumnale) принадлежит к семейству лилейных, цветет на лугах поздним летом и осенью, листья и семена появляются весной следующего года. Все части растения содержат алкалоид колхицин. Это вещество тормозит полимеризацию тубулина, приводящую к формированию микротрубочек, ответственных за внутриклеточный транспорт. Под влиянием колхицина макрофаги и нейтрофилы теряют способность к внутриклеточному транспорту органелл. На этом принципе основано лечение острых приступов подагры. Колхицин блокирует митоз на стадии метафазы, нарушая образование веретена.
Разработка фармакологических препаратов [ править | править код ]
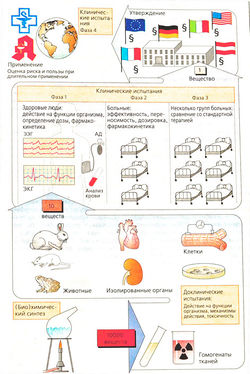
Разработка новых лекарств основана на синтезе новых химических соединений. Вещества сложного строения получают из растений (например, сердечные гликозиды), тканей животных (гепарин), на основе культур микроорганизмов (пенициллин) или культур клеток человека (урокиназа), а также с помощью генной инженерии (человеческий инсулин). Чем больше известно о связи между химическим строением вещества и его воздействием, тем целенаправленнее осуществляется поиск нового препарата.
Действие нового вещества проверяется при доклиническом исследовании. На начальной стадии проводят фармакобиохимические исследования (например, эксперименты по связыванию с рецепторами) или изучение на клеточных культурах, изолированных клетках или органах. Однако, поскольку в таких модельных исследованиях нельзя воспроизвести сложные биохимические процессы, происходящие в живом организме, дальнейшие испытания нового препарата проводятся на животных. Эксперименты на животных показывают, имеет ли препарат лечебное действие, и какова вероятность токсического влияния.
Токсикологические исследования проводятся для определения токсичности при кратковременном или длительном применении (острая и хроническая токсичность), способности повреждать генетическую структуру (мутагенность), способствовать возникновению опухолей (канцерогенность) или нарушать развитие плода (тератогенность). В экспериментах на животных изучают, как видоизменяется препарат при всасывании, распределении и выделении (фармакокинетика).
Лишь небольшая часть соединений выдерживает доклинические испытания и применяется в дальнейшем для лечения людей. Фармацевтическая технология предлагает для найденного вещества лекарственную форму.
Фаза 1 клинических испытаний проводится на людях — здоровых добровольцах — для проверки действия, выявленного в экспериментах на животных. Необходимо установить зависимость действия лекарства от его дозы. В фазе 2 клинических испытаний препарат назначают небольшому числу больных. При положительных результатах лечения и незначительных побочных эффектах проводится фаза 3 клинических испытаний уже на большем числе пациентов. При этом новое вещество сравнивается с уже применяемыми лекарствами. При клинических испытаниях многие из препаратов признаются негодными к применению.
Из 10 000 вновь синтезированных соединений лишь одно вещество становится лекарством.
Решение о допущении лекарства к применению принимается государственными учреждениями (BfArM — Bundesinstitut fur Arzneimittel und Medizinprodukte в Бонне; EMEA — European Agency for the Evaluation of Medicinal Products в Лондоне). Заявитель нового лекарства обязан предъявить результаты всех испытаний и их соответствие предъявляемым критериям фармакологической активности и качеству лекарственной формы.
После получения разрешения и присвоения коммерческого названия новое лекарство поступает в продажу и может быть назначено врачом и выдано аптекой. Наблюдение за действием используемого препарата продолжается и далее (фаза 4 клинических испытаний). Лишь после многолетнего применения препарата и оценки побочного действия делается заключение о терапевтическом значении нового лекарства. Если у нового лекарственного препарата нет явных преимуществ, учитывают соотношение его стоимости и спроса.
Источник
Этапы разработки лекарственных препаратов
Этапы разработки лекарственных средств [ править | править код ]
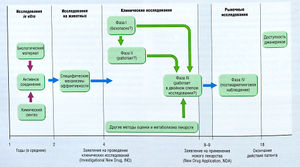
Между моментами создания нового лекарства и демонстрации его клинической эффективности и адекватной безопасности можно выделить несколько этапов (рис. 3.1). Этап первоначальной разработки обычно состоит в определении терапевтической дели (заболевание или состояние) или целевой молекулы, например рецептора, фермента и др., и последующем обнаружении основного химического соединения, т.е. вещества с характерным эффектом, необходимым для нового лекарства. В современных программах разработки лекарств чаще определяется целевая молекула, которая является ключевым звеном патологического процесса, и осуществляется поиск синтетических и природных соединений, действующих на эту молекулу. В дальнейшем пытаются разработать более подходящие соединения. Получение таких соединений — многократный процесс, включающий синтез похожих химических производных основного соединения. При разработке новых аналогов, чтобы получить требуемую эффективность, используют анализ взаимосвязи структура-активность (SAR или QSAR при количественной оценке).
Описание к рис. 3.1 Процесс разработки и оценки лекарства для вывода на рынок США. Некоторые требования для препаратов, используемых для лечения жизнеугрожающих заболеваний, могут отличаться [Katzung ВС. Basic and Clinical Pharmacology, 6th ed. New York: I Appleton & Lange]
Некоторые аналоги препаратов становятся объектами крупномасштабных фармакологических и токсикологических исследований для определения характеристик лекарств, которые могут получить одобрение для клинических исследований с участием пациентов. После серии клинических наблюдений полученные данные подаются в регулирующий орган для получения разрешения на реализацию нового лекарства. После этого с помощью различных методов собирают результаты клинического применения препарата. Этот процесс называют постмаркетинговыми наблюдениями (см. Принятие фармакотерапевтического решения), которые регулируют менее строго, чем процедуры, необходимые до получения регистрации.
Эксперименты на животных обеспечивают основу клинических наблюдений
Сведения о фармакологических эффектах лекарства in vitro и in vivo используют для предварительного заключения о его терапевтической ценности. Эти данные нужны для обоснования исследований на людях, поскольку без них не будет базы для оценки ожидаемой пользы и приемлемого риска нежелательных эффектов. Доклиническими исследованиями называют эксперименты in vitro и на животных, используемые для определения действия лекарства на уровне молекулы, клетки, определенной ткани или органа, оценки фармакологических свойств и изучения потенциальных терапевтических эффектов на животных моделях заболеваний человека. Исследования на животных также помогают изучить метаболизм и распределение лекарства в организме и разработать основные показания. Клинические исследования не могут быть продолжены, если не доказана безопасность лекарства. Для оценки возможной токсичности нового лекарства необходимы следующие исследования на животных:
- токсикологические исследования in vitro для оценки генетической и биохимической токсичности;
- оценка острой токсичности с изучением физиологических систем (сердечно-сосудистая, центральная нервная, желудочно-кишечный тракт), кожи и слизистых (острое раздражение и возбуждение);
- оценка подострой и хронической токсичности;
- оценка канцерогенности;
- оценка репродуктивной токсичности;
- оценка генетической токсичности.
При изучении острой токсичности оценивают эффекты, возникающие через несколько часов или дней после однократного введения. При изучении хронической токсичности рассматривают эффекты после введения повторных доз в течение нескольких недель или месяцев.
Однако надежность данных, полученных на животных, для прогнозирования клинических результатов зависит от уровня клинической релевантности модели. Например, модель пневмонии, вызванной золотистым стафилококком, хорошо прогнозируема. Инфицирование организма одинаково и у людей, и у животных. Иммунологический ответ против бактерий и легочная патология у животных и человека очень схожи. Напротив, животные модели других заболеваний только косвенно имитируют заболевания человека и менее предсказуемы. Обычно возможность разработки животной модели связана с пониманием патофизиологии конкретного заболевания. В указанном примере непосредственная причина пневмонии хорошо известна, в то время как точная этиология многих заболеваний не определена.
Изучение лекарства в клинике состоит из нескольких этапов
Клинические исследования начинаются после того, как собрано достаточное количество данных после исследований на животных в качестве обоснования для оценки нового лекарства в клинике и получения необходимого официального разрешения. Этапы разработки лекарства обозначают как фаза I, фаза II и фаза III. Фаза IV является этапом пост-маркетинговых наблюдений и других пострегистрационных клинических исследований (см. рис. 3.1).
Фаза I включает первые клинические исследования с участием людей. Эти исследования проводят под очень строгим наблюдением, обычно они являются открытыми или одинарными слепыми (табл.3.2) и определяют наименьшую допустимую дозу по токсичности. Дальнейшие исследования проводят с меньшими дозами. Обычно в таких исследованиях участвуют молодые здоровые мужчины. В дальнейшем их заменяют группой больных. Также в эту фазу получают первичные данные о фармакокинетике.
Фаза II начинается после определения диапазона допустимых доз и рассматривается как доказательство концепции. Этот этап проходит с участием больных, у которых новое лекарство должно проявить свой потенциальный эффект. Основная цель состоит в получении доказательств того, что новое лекарство эффективно, т.е. обладает эффектами, полученными в доклинических исследованиях. Иногда конечной точкой клинических наблюдений фазы II является собственно терапия, в других случаях используют заместительные конечные точки исследований. Заместительная конечная точка прогнозирует или предположительно прогнозирует истинную конечную точку. Например, изучение лекарства при сердечной недостаточности может иметь истинную конечную точку при увеличении толерантности к нагрузке или выживаемости. Заместительная конечная точка для того же лекарства может быть уменьшением периферического сопротивления сосудов и улучшением сердечного выброса. Для лекарства, которое может предотвращать тромбообразование при ангиопластике, заместительной конечной точкой может быть ингибирование агрегации тромбоцитов, а истинной конечной точкой — уменьшение рестеноза.
Заместительная конечная точка наиболее удобна, когда она тесно связана с истинной конечной точкой. Так, например, заместительной конечной точкой является снижение артериального давления. Целью лечения гипертензии является снижение неблагоприятных сердечно-сосудистых реакций организма и почечной недостаточности как последствий гипертензии. Таким образом, снижение артериального давления — это заместительная конечная точка для уменьшения последствий гипертензии.
Другие цели фазы II состоят в определении фармакокинетики лекарства и связи между эффектом и концентрацией вещества в плазме, если это возможно. Также изучается влияние заболеваний печени и почек на выведение лекарства из организма, фармакокинетические и фармакодинамические взаимодействия нового лекарства с другими средствами, с которыми их могут назначать совместно.
Исследования в фазу II могут быть одинарными или двойными слепыми, параллельными или перекрестными, с использованием случайных выборок пациентов. В этнически разнородных популяциях, например в США, в фармакокинетических исследованиях иногда изучают особенности метаболизма лекарств у разных этнических групп. Этническая однородность является грубым усреднением генетической классификации. Возможно, в будущем более корректный подход к оценке путей метаболизма и клинических результатов будет состоять в классификации пациентов по их генетической предрасположенности к метаболизму лекарств. Тогда будет возможно предсказать, для какого генотипа лекарство будет более полезно, а для какого — токсично. Этот раздел фармакологии называют фармакогенетикой.
В фазу III устанавливают эффективность и безопасность нового лекарства. Если возможно, проводят контролируемые рандомизированные двойные слепые исследования, которые всегда параллельны. Планируемая модель и размер всех клинических наблюдений, особенно фазы III, основывают на статистических действиях, например рандомизации процедур, чтобы после окончания исследования получить веское заключение. Кроме того, популяционные исследования фазы III должны усреднять целевую популяцию для данного лекарства. В исследовании должны участвовать пациенты с различными проявлениями изучаемого заболевания. Распределение по этническим группам и полу должно отражать таковое в популяции. Наибольшее внимание уделяют изучению детей, за исключением случаев, когда это нецелесообразно, например при изучении лекарств для лечения таких заболеваний у пожилых, как болезнь Альцгеймера.
Разработка лекарств является длительным процессом
- Время от подачи заявки на регистрацию до его получения составляет от 6 мес до нескольких лет, чаще 1-2 года
- Процесс разработки лекарства до регистрации обычно занимает 6-10 лет
Таблица 3.2 Клинические исследования, терминология
Стандартная терапия (или плацебо при отсутствии стандартов), с которой сравнивают эффективность нового препарата
Пациенты, участвующие в исследовании, имеют одинаковую возможность быть включенными в опытную или контрольную группу, а факторы, которые могут повлиять на результаты, одинаково распределены между двумя группами
Двойное слепое исследование
Ни врач, ни пациент не знают, получает ли данный пациент опытное или контрольное средство, что помогает избежать субъективизма
Одинарное слепое исследование
Врач знает, какой препарат назначен данному пациенту, но пациент не знает
Противоположно двойному слепому: и врач, и пациент знают, какое средство (опытное или контрольное)назначено и в какой дозе
Одновременно оценивают как минимум две схемы, но пациенту назначают только один вид терапии
Пациенты получают каждый вид лечения последовательно и таким образом выступают в качестве контрольной группы для самих себя. Например, если лечение А оценивают относительно лечения В, то некоторые пациенты получают сначала А, потом В, а другие наоборот — сначала В, потом А. Так оценивают эффекты лекарственной терапии, а не порядка назначений
Измеряют для оценки эффекта лекарства (например, нормализация артериального давления — конечная точка для оценки антигипертензивных средств, уменьшение боли — конечная точка для оценки анальгетиков)
Заместительная конечная точка
Результат лечения, который прогнозирует истинную цель терапии, не являясь этой целью (например, снижение размера опухоли в качестве заместителя выживаемости)
Источник


