- Сертификат GMP: подтверждение качества лекарственных средств
- К каким производствам применима эта процедура?
- Нормативная база
- Преимущества обладания сертификатом
- Стандарт GMP в международной практике
- Правила GMP в России
- Процедура получения сертификата в России
- Документы для сертификации
- Сроки сертификации
- Стоимость получения сертификата
- Национальный стандарт РФ ГОСТ Р 52249-2009 «Правила производства и контроля качества лекарственных средств» (утв. и введен в действие приказом Федерального агентства по техническому регулированию и метрологии от 20 мая 2009 г. N 159-ст)
- ГАРАНТ:
Сертификат GMP: подтверждение качества лекарственных средств

Сертификат GMP — это соблюдение изготовителем лекарственных препаратов требований надлежащей производственной практики. В России они сформулированы в национальном стандарте ГОСТ Р 52249-2009, который идентичен правилам, действующим в Европейском Союзе.
К каким производствам применима эта процедура?
В настоящее время в странах, которые контролируют соответствие стандарту GMP на своих территориях, его правила применяются для проверки качества следующих категорий продукции:
- лекарственные препараты;
- медицинские изделия различного назначения, включая те из них, которые применяются в диагностических целях;
- продукты питания и ингредиенты для их производства;
- биологически активные добавки.
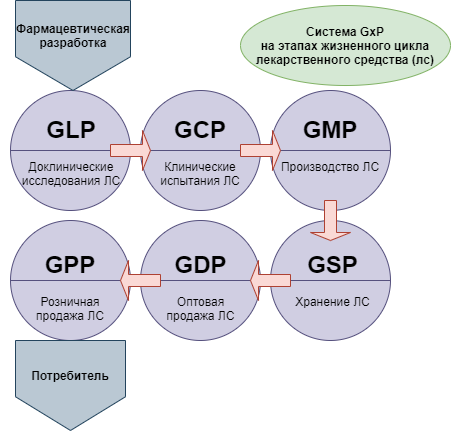
При этом для понимания ситуации следует принимать во внимание, что новая версия сертификации GMP — это не единственная система требований, которые в международной практике применяются в целях стандартизации медицинского обслуживания населения. Кроме них, производителям, работающим в такой сфере как фармация, необходимо соответствовать требованиям комплекса правил, объединенных под общим наименованием GxP:
- GLP — Good Laboratory Practice (надлежащая лабораторная практика);
- GCP — Good Clinical Practice (надлежащая клиническая практика);
- GDP — Good Distributon Practice (надлежащая дистрибьюторская практика);
- GACP — Good Agricultural and Collection Practice (надлежащая практика культивирования и сбора лекарственных растений).
Нормативная база
В Российской Федерации получение сертификата GMP осуществляется на основании действующей нормативной базы, включающей следующие основные правовые акты:
- национальный стандарт РФ ГОСТ Р 52249-2009, устанавливающий правила изготовления и контроля качества лекарственных препаратов;
- постановление Правительства от 5 июня 2008 года N 438 с рядом изменений, внесенных за последние годы, которое утверждает полномочия Министерства промышленности и торговли в этой области;
- постановление Правительства от 3 декабря 2015 года N 1314, устанавливающее порядок оценки соответствия производителей требованиям стандарта надлежащей практики;
- приказ Минпромторга от 14 июня 2013 года N 916, утверждающий правила применения надлежащей производственной практики в соответствии с актуальным стандартом;
- приказ Минпромторга от 26 мая 2016 года N 1714, определяющий административный регламент предоставления государственной услуги по выдаче документации, подтверждающей соответствие изготовителя установленным нормам надлежащей производственной практики;
- приказ Минпромторга России от 17.12.2015 N 4119, утверждающий правила ведения реестра сведений о том, какие лекарства имеют сертификат качества GMP в России.
При этом необходимо принимать во внимание, что в настоящий момент наша страна вместе с другими государствами, входящими в состав Евразийского экономического союза, находится на этапе становления общего рынка, объединяющего фармацевтическое и косметическое производство в границах Союза. Это предполагает в том числе введение в действие единых требований к качеству и безопасности таких продуктов. В соответствии с принятым в мире порядком они реализуются в форме внедрения стандартов надлежащей производственной практики. Применение таких стандартов регулируется следующими нормативными документами:
- Решение Совета ЕЭК от 3 ноября 2016 года N 77, утверждающее правила надлежащей производственной практики в границах ЕАЭС;
- Приказ Минпромторга от 4 сентября 2020 года N 2945, которым введен административный регламент предоставления госуслуги по выдаче документации, подтверждающей соответствие производств установленным правилам.
Обратите внимание!
Для полноценного применения разработанного административного регламента необходимо решение Правительства о порядке реализации некоторых процедур, связанных с проведением фармацевтических инспекций. Приказ № 2945 вступит в силу только после принятия соответствующего постановления: пока этого не произошло.
Преимущества обладания сертификатом
Несмотря на необходимость проведения достаточно сложной и дорогостоящей процедуры, производители знают, что сертификация по стандартам GMP является весьма важной для представителей фармацевтической отрасли. В частности, оно обеспечивает продукции и производству следующие серьезные преимущества:
- стабильное качество продукции, не зависящее от внешних факторов;
- повышение доверия потребителей, включая крупных оптовых покупателей, которые всегда отслеживают, какие производители имеют сертификат соответствия GMP на их продукцию;
- возможность вывода продукции на международные рынки, где ее может купить гораздо больше потребителей;
- возможность привлечения инвесторов для реализации проектов по расширению производства;
- получение преимуществ при участии в конкурсном отборе поставщиков, в том числе для государственных закупок.
КОММЕНТАРИЙ ЭКСПЕРТА АТТЭК
Каков срок действия сертификата?
Срок действия российских сертификатов составляет 3 года. При этом срок действия иностранного сертификата GMP составляет от 1 до 3 лет. По истечении этого периода сертификацию потребуется проходить заново. Кроме того, это означает, что на протяжении всего этого срока компании необходимо обеспечить соответствие своего производства и продукции требованиям комплекса правил GMP.
Кто в России занимается сертификацией по стандартам GMP?
Сейчас сертификация контролируется департаментом развития медицинской и фармацевтической промышленности Министерства промышленности и торговли РФ. Он является организацией, ответственной за обеспечение надлежащего контроля за качеством, безопасностью и эффективностью лекарственных средств. Осуществлением требуемых сертификационных процедур занимается Государственный институт лекарственных средств и надлежащих практик (ФГБУ «ГИЛС и НП»).
Стандарт GMP в международной практике
Процесс сертификации на соответствие лекарственного препарата стандартам GMP в международной практике имеет комплексный характер, а ее основной целью является подтверждение безопасности и действенности продукции. В этой связи для достижения поставленной цели специалисты аккредитованных сертификационных организаций не ограничиваются оценкой ряда выборочных образцов лекарственных препаратов, как это часто предусматривается другими стандартами. В процедуру установления требуемого уровня качества лекарств любой международный центр сертификации лекарственных средств включает оценку предприятия, занимающегося его выпуском. В результате эксперты, занимающиеся проведением сертификации, анализируют конкретный препарат и процесс его выпуска в следующих областях:
- оценка производства на соответствие критериям безопасности, включая проведение его проверки в отношении вероятности попадания в продукт посторонних примесей и веществ;
- оценка производства на соблюдение технических требований к выпуску продукции, включая выполнение условий относительно влажности, температуры и других параметров в производственных помещениях;
- оценка качества, безопасности и действенности лекарственных средств, производимых на конкретном предприятии;
- оценка соответствия параметров производства и характеристик лекарственного средства нормативной документации, принятой в рамках процедуры GMP.
Правила GMP в России
Порядок и сроки проведения всех операций в рамках этой процедуры, список лиц и организаций, ответственных за их осуществление, размер платы за проведение экспертной оценки и другие аспекты выполнения сертификации определены постановлением Правительства № 1314.
Процедура получения сертификата в России
Первым шагом для производителя, который желает пройти сертификацию, является подача соответствующего заявления в Минпромторг. В течение 10 рабочих дней специалисты ведомства проводят проверку корректности представленных в заявлении сведений и определяют возможность проведения сертификации.
В случае необходимости они вправе запросить у заявителя дополнительные документы, которые он обязан предоставить в течение 20 рабочих дней. В случае, если в отношении данного препарата принято положительное решение о проведении процедуры сертификации, необходимые данные направляются в ФГБУ «ГИЛС и НП», который в течение 20 рабочих дней с момента их получения обязан определить дату проведения сертификационных мероприятий и внести ее в график. Такая дата должна наступить не позднее 160 рабочих дней со дня, когда специалисты Минпромторга приняли положительное решение о сертификации, а сама экспертиза и расшифровка ее результатов должны занимать не более 10 рабочих дней.
На подготовку итогового отчета по результатам ее проведения исполнителю отводится 30 рабочих дней, а на его направление заявителю — 3 рабочих дня. Копия такого отчета также направляется в Минпромторг. На основании отчета формируется окончательное заключение, которое в случае положительного характера сопровождается выдачей сертификата производителю лекарственного препарата.
Документы для сертификации
Чтобы получить сертификат GMP в России, производитель обращается в уполномоченный орган с заявлением, к которому прилагает пакет документов, включающий:
- копию документа, подтверждающего наличие у заявителя полномочий по взаимодействию с контролирующей организацией;
- копия основного досье используемого производственного объекта;
- информация о фактах несоответствия препарата действующим требованиям к качеству и безопасности и о фактах отзыва медикамента из оборота за период не менее 2 лет;
- полный список лекарств, который изготавливаются на данном производственном объекте;
- копия лицензии на производство лекарств;
- письмо о согласии на проведение инспекции производства.
Важнейшие документы предоставляются заявителем в копиях, поскольку при утере их восстановить невозможно или очень сложно. Правила регламентируют, что если заявление подает иностранный производитель, и некоторые документы в составе пакета представлены на другом языке, они должны быть переведены на русский язык и заверены в установленном порядке.
Сроки сертификации
Общая продолжительность процедуры сертификации складывается из следующих сроков.
Этап сертификационной процедуры
Максимальная допустимая продолжительность
Проверка полноты пакета документации, представленной с заявлением о сертификации, и правильности ее оформления, назначение инспекции
10 рабочих дней
Направление информации о назначении инспектирования в уполномоченное учреждение, которое проводит проверку
Инспектирование и анализ лекарственного средства
160 рабочих дней
Принятие решения о выдаче заключения по результатам инспекционного отчета
10 рабочих дней
160-дневный период инспектирования включает внесение производителя в график инспекций, ожидание процедуры и проведение самой инспекции. Она должна занимать не более 10 рабочих дней.
Такой порядок действует, если в документации, поданной производителем, не обнаружат ошибок и недочетов, из-за которых ее могут направить на доработку. В этом случае вся процедура займет немногим более 180 рабочих дней, то есть свыше 8 месяцев.
Стоимость получения сертификата
Обязательной для всех производителей лекарственных средств, претендующих на получение сертификата, подтверждающего соответствие их продукции стандартам GMP, является оплата государственной пошлины за рассмотрение соответствующего заявления в Министерстве промышленности и торговли. Ее размер составляет 7500 рублей. Оплатить данную сумму необходимо еще до подачи заявления в ведомство, а ее размер никак не зависит от результатов рассмотрения документа.
Однако данная пошлина — это далеко не единственный и не самый крупный платеж, который потребуется осуществить производителю лекарств. Другой значительной статьей расходов станет плата за проведение экспертной оценки производства и продукции заявителя. Такая процедура выполняется специалистами ФГБУ «ГИЛС и НП»: для каждого из них предварительно проводится аттестация эксперта по GMP в России.
При этом размер платы за проведение оценки не является строго установленным, а определяется в зависимости от объема, характера и сложности необходимых процедур в соответствии с положениями приказа Министерства промышленности и торговли Российской Федерации от 11.01.2016 № 9 «Об утверждении методики определения размера платы за оказание услуги по инспектированию GMP». В случае, если проверка потребует проведения значительного объема работы и привлечения большого количества высококвалифицированных экспертов, размер платы за ее проведение может превышать 2,5 миллиона рублей.
Источник
Национальный стандарт РФ ГОСТ Р 52249-2009 «Правила производства и контроля качества лекарственных средств» (утв. и введен в действие приказом Федерального агентства по техническому регулированию и метрологии от 20 мая 2009 г. N 159-ст)
Национальный стандарт РФ ГОСТ Р 52249-2009
«Правила производства и контроля качества лекарственных средств»
(утв. и введен в действие приказом Федерального агентства по техническому регулированию и метрологии от 20 мая 2009 г. N 159-ст)
Good manufacturing practice for medicinal products (GMP)
Дата введения — 1 января 2010 г.
Взамен ГОСТ Р 52249-2004
ГАРАНТ:
См. также Правила организации производства и контроля качества лекарственных средств, утвержденные приказом Минпромторга России от 14 июня 2013 г. N 916
О производстве лекарственных средств см. Федеральный закон от 12 апреля 2010 г. N 61-ФЗ
Цели и принципы стандартизации в Российской Федерации установлены Федеральным законом от 27 декабря 2002 г. N 184-ФЗ «О техническом регулировании», а правила применения национальных стандартов Российской Федерации — ГОСТ Р 1.0-2004 «Стандартизация в Российской Федерации. Основные положения»
Настоящий стандарт является идентичным переводом правил GMP Европейского Союза (GMP ЕС) «Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use» no состоянию на 31 января 2009 г.
Впервые правила GMP ЕС были приняты в России в 2004 г. в качестве ГОСТ Р 52249-2004 «Правила производства и контроля качества лекарственных средств», который соответствовал правилам GMP ЕС 2003 г. За истекшее время в правила GMP ЕС внесены следующие существенные изменения и дополнения:
— добавлены новые требования в основной текст;
— внесены изменения в:
— приложение 1 Производство стерильных лекарственных средств;
— приложение 3 Производство радиофармацевтических препаратов;
— приложение 7 Производство лекарственных средств из растительного сырья;
— приложение 13 Производство лекарственных средств для исследований;
— введены два новых приложения:
— приложение 19 Контрольные и архивные образцы;
— приложение 20 Анализ рисков для качества;
— внесен ряд других изменений.
Также была изменена структура данного стандарта. В ГОСТ Р 52249-2004 приложение 18 содержало «Руководство по производству активных фармацевтических субстанций (АФС)», что соответствовало Правилам GMP ЕС на 2003 г. В новой редакции правил GMP ЕС требования к производству АФС перенесены из приложения в основную часть, которая содержит теперь две части:
— часть I Основные требования и #
— часть II Основные требования к активным фармацевтическим субстанциям (АФС), используемым в качестве исходных материалов.
Приложение 18 отсутствует, хотя сохранен его номер. Такой порядок структуры стандарта сохранен и в ГОСТ Р 52249-2009.
В последнюю редакцию правил GMP ЕС введено приложение 20 об анализе рисков, являющееся текстом руководства ICH Q9 «Анализ рисков для качества» (Quality Risk Management). Этот текст изложен неконкретно и непригоден к практическому применению. В связи с этим приложение 20 в текст нового стандарта не включено, о чем указано в сносках в тексте стандарта, выделенных курсивом.
По тексту стандарта опущены ссылки на Директивы ЕС, содержащиеся в оригинале правил GMP ЕС.
Настоящий стандарт устанавливает требования к производству и контролю качества лекарственных средств для человека и животных.
Стандарт распространяется на все виды лекарственных средств и устанавливает общие требования к их производству и контролю качества, а также специальные требования к производству активных фармацевтических субстанций (часть II) и отдельных видов лекарственных средств (приложения 1 — 19),
Стандарт не устанавливает требований к обеспечению промышленной безопасности, пожарной безопасности, взрывобезопасности, химической безопасности и безопасности других видов при производстве лекарственных средств.
*(1) В данном разделе правил GMP ЕС дана ссылка на приложение 20 по методам анализа рисков. В настоящий стандарт приложение 20 не включено ввиду его неконкретности и резкой критики в его адрес со стороны специалистов.
*(2) За исключением методов, установленных нормативными документами.
*(3) ICH — International Conference for Harmonization (Международная конференция по гармонизации). Здесь и далее по тексту.
*(4) Руководство ICH Q5A «Качество биотехнологических продуктов. Оценка безопасности вирусного заражения биотехнологических продуктов, произведенных из клеток человеческого и животного происхождения» (Quality of Biotechnological Products: Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin).
*(5) Руководство ICH Q5A «Качество биотехнологических продуктов. Выделение и характеристика клеточных субстратов, используемых для производства биотехнологических/биологических продуктов» (Quality of Biotechnological Products; Derivation and Characterization of Cell Substrates Used for Production of Biotechnological/ Biological Products).
Общие термины и определения
В настоящем стандарте используются следующие термины с соответствующими определениями:
1 аттестация; испытания (qualification, validation): Доказательство того, что методика, процесс, оборудование, материал, операция или система соответствуют заданным требованиям и их использование действительно дает ожидаемые результаты.
2 баллон (cylinder): Сосуд для хранения газа при высоком давлении.
3 банки клеток (cell bank) :
система банков клеток (cell bank system): Система, при которой последовательные серии продукции производят из клеточных культур, принадлежащих главному банку клеток, который полностью охарактеризован на подлинность и отсутствие загрязнений. Некоторое количество емкостей главного банка клеток используется для формирования рабочего банка клеток. Система банков клеток должна быть аттестована (испытана) на определенное количество пересевов или количество удвоений популяции, до достижения которых они могут использоваться в текущем производстве.
главный банк клеток (master cell bank): Культура клеток, полностью охарактеризованная на подлинность и отсутствие загрязнений, распределенная по емкостям в процессе одной операции таким образом, чтобы обеспечивались ее однородность и стабильность. Как правило, главный банк клеток хранится при температуре минус 70°С или ниже.
рабочий банк клеток (working cell bank): Культура клеток, отобранная из главного банка клеток для приготовления производственных культур клеток. Рабочие банки клеток хранят при температуре минус 70°С или ниже.
4 биологические агенты (biological agents): Микроорганизмы, в т.ч. полученные методами генной инженерии, культуры клеток и эндопаразиты, патогенные или непатогенные.
5 биореактор (biogenerator): Изолированная система (например, ферментатор), в которую вместе с другими материалами вводят биологические агенты и в результате протекающей реакции происходит их размножение или образование других веществ. Биореакторы, как правило, оборудуются устройствами для регулирования, контроля, добавления и извлечения материалов.
6 внутрипроизводственный контроль (in-process control) : Контроль, выполняемый в ходе технологического процесса с целью проверки соответствия продукции заданным требованиям, по результатам которого может выполняться корректировка параметров технологического процесса. Контроль состояния окружающей среды или оборудования рассматривается как элемент внутрипроизводственного контроля.
7 возврат (return): Возврат лекарственного средства его производителю или поставщику.
8 воздушный шлюз (air-lock): Ограниченное пространство с двумя или несколькими дверями между двумя или несколькими помещениями (например, различных классов чистоты), предназначенное для разделения воздушных сред помещений при входе в них. Воздушный шлюз служит для перехода персонала или перемещения материалов.
9 готовая продукция (готовый продукт) (finished product): Лекарственное средство, прошедшее все этапы технологического процесса, в т.ч. окончательную упаковку.
10 изолированная зона (contained area): Зона, оборудованная соответствующими фильтрами и устройствами подготовки воздуха для предотвращения загрязнения внешней окружающей среды биологическими агентами, присутствующими в этой зоне.
11 изоляция (containment): Меры по ограничению распространения биологического агента или другого вещества за пределы определенного пространства.
первичная изоляция : Изоляция, препятствующая прониканию биологического агента в окружающую среду, непосредственно прилегающую к рабочей зоне. Предусматривает использование закрытых контейнеров или безопасных биологических шкафов и методов безопасного выполнения технологических операций.
вторичная изоляция : Изоляция, препятствующая прониканию биологических агентов во внешнюю окружающую среду или в другие рабочие зоны. Предусматривает использование помещений со специальными системами подготовки воздуха, воздушных шлюзов и/или стерилизаторов для перемещения материалов и обеспечивает безопасное выполнение технологических операций. Во многих случаях используется для повышения эффективности первичной изоляции.
12 инструкция; методика; процедура (procedure): Документ, содержащий указания по выполнению отдельных видов операций (например, по очистке, переодеванию, контролю окружающей среды, отбору проб, проведению испытаний, эксплуатации оборудования).
13 инфицированный (infected): Зараженный чужеродными биологическими агентами и способный к распространению инфекции.
14 испытания (validation) : См. аттестация.
15 исходные материалы (starting material): Любое вещество, используемое для производства лекарственных средств, кроме упаковочных материалов.
16 калибровка; поверка (calibration): Операции, устанавливающие при определенных условиях зависимость между значениями, регистрируемыми контрольно-измерительными приборами (системами) и соответствующими стандартными величинами (эталонами).
17 карантин (quarantine): Статус исходных или упаковочных материалов, промежуточной, нерасфасованной или готовой продукции, изолированных физически или иным образом до вынесения решения об их выпуске или отклонении.
18 коллектор (manifold): Устройство или оборудование, позволяющее одновременно наполнять газом несколько баллонов (контейнеров) из одного источника.
19 контролируемая зона (controlled area): Зона, построенная и эксплуатируемая таким образом, чтобы предотвратить внесение возможного загрязнения и случайное распространение живых организмов (например, может иметь систему воздухоподготовки, соответствующую зоне D). Степень контроля зависит от свойств организмов, используемых в технологическом процессе. Как минимум, такая зона должна иметь отрицательное давление по отношению к смежным помещениям и возможность эффективно удалять незначительные количества аэрозольных загрязнений.
20 контроль качества (quality control): См. пункт 1.4 раздела 1.
21 криогенный сосуд (cryogenic vessel): Сосуд для хранения сжиженных газов при сверхнизких температурах.
22 культура клеток (cell culture): Клеточная масса, полученная в результате выращивания in vitro клеток, изолированных от многоклеточных организмов.
23 лекарственное растение (medicinal plant): Растение или часть его, используемое для медицинских целей.
24 лекарственное средство (medicinal product): Любое вещество или комбинация веществ, предназначенное для лечения или профилактики заболеваний у человека или животных. Любое вещество или комбинация веществ, вводимое человеку или животным с диагностическими целями для восстановления, корректировки или изменения физиологических функций человека или животных, также рассматривается как лекарственное средство.
25 лекарственное средство из растительного сырья (herbal medicinal product): Лекарственное средство, содержащее в качестве активных ингредиентов только вещества растительного происхождения и/или препараты на их основе.
26 нерасфасованный готовый продукт; балк-продукт (bulk product): Продукт, прошедший все производственные стадии, за исключением окончательной упаковки.
27 номер серии; номер партии (batch number or lot number): Определенное сочетание цифр и/или букв, обозначающее серию продукции.
28 перекрестное загрязнение (cross contamination): Загрязнение материалов или продукции другими материалами или продукцией.
29 переработка (reprocessing): Повторная обработка серии или части серии продукции, не соответствующей заданным требованиям, начиная с определенной стадии производства, для получения продукции требуемого качества после проведения одной или нескольких дополнительных операций.
30 повторное использование (recovery): Включение произведенной ранее серии продукции требуемого качества (или ее части) в другую серию продукции на определенной стадии производства.
31 посевной материал (seed lot):
система посевных материалов (seed lot system): Система, при которой последовательные серии продукции получают из главного посевного материала при определенном количестве пересевов (пассажей). В текущем производстве рабочий посевной материал готовится из главного посевного материала. Готовый продукт производится из рабочего посевного материала, при этом число пересевов из главного посевного материала не должно превышать значения, установленного при клинических испытаниях вакцин, исходя из требований безопасности и эффективности. Происхождение главного посевного материала и история пересевов из него должны оформляться документально.
главный посевной материал (master seed lot): Культура микроорганизмов, распределенная из одного нерасфасованного продукта по емкостям в процессе одной операции таким образом, чтобы обеспечивались однородность, стабильность и не допускалось загрязнение. Главный посевной материал в жидком виде хранят при температуре минус 70°С; в лиофилизированном виде — при температуре, обеспечивающей его стабильность.
рабочий посевной материал (working seed lot): Культура микроорганизмов, полученная из главного посевного материала и предназначенная для использования в производстве. Рабочий посевной материал распределяется по емкостям и хранится аналогично хранению главного посевного материала.
32 производитель (manufacturer): Держатель лицензии на производство.
Примечание — Предприятие, осуществляющее хотя бы один этап производства, рассматривается как производитель лекарственных средств.
33 производство (manufacture): Все операции и виды контроля, связанные с получением, приемкой и обработкой исходных материалов, упаковкой, выпуском в реализацию, хранением и отгрузкой лекарственных средств.
34 промежуточный продукт (intermediate product): Частично обработанный материал, который должен пройти дальнейшие стадии производства, прежде чем он станет нерасфасованным готовым продуктом.
35 промышленный регламент, технологические инструкции и инструкции по упаковке (manufacturing formulae, processing and packaging instructions): Документы, определяющие все используемые исходные материалы и операции по производству и упаковке продукции.
1 Промышленный регламент является технологическим документом действующего серийного производства, устанавливающим методы производства, технологические нормативы, средства, условия и порядок проведения технологического процесса и обеспечивающим получение лекарственного средства с показателями качества, отвечающими требованиям нормативной документации.
2 Данный термин используется только в области производства лекарственных средств.
3 Промышленный регламент не относится к техническим регламентам, предусмотренным Федеральным законом «О техническом регулировании».
36 протокол на серию (record): Документ, отражающий процесс производства каждой серии продукции, в т.ч. разрешение на ее реализацию, и все факторы, имеющие отношение к качеству готовой продукции.
37 радиофармацевтический препарат (radiopharmaceutical): Любое лекарственное средство, содержащее в готовом виде один или более радионуклидов (радиоактивных изотопов), используемых для медицинских целей.
38 растительное сырье (crude plant; vegetable drug): Сырые или высушенные лекарственные растения или их части.
39 регистрационное досье (registration dossier): Комплект документов и материалов установленной структуры и содержания, представляемый вместе с заявкой на регистрацию лекарственного средства, а также утвержденный в процессе регистрации.
40 серия; партия (batch; or lot): Определенное количество однородных исходных и упаковочных материалов или однородной продукции, обработанной в ходе одной или нескольких последовательных технологических стадий.
Примечание — При необходимости на определенных стадиях производства серия может быть разделена на подсерии, объединяемые впоследствии в однородную серию продукции. При непрерывном производстве понятие серии должно относиться к определенной части продукции, характеризуемой однородностью.
С точки зрения контроля готовой продукции серия продукции включает в себя совокупность единиц дозированной формы лекарственных средств (лекарственной формы), изготовленных из одного объема исходного материала и прошедших единую последовательность производственных операций или единый цикл стерилизации; при непрерывном производстве — все единицы, произведенные в заданный интервал времени.
41 сжиженные газы (liquifiable gases): Газы, которые при стандартных температуре и давлении наполнения находятся в баллоне в сжиженном виде.
42 система (system): Совокупность взаимосвязанных действий и технических средств, образующих единое целое.
43 система с компьютерным управлением и контролем (computerized system): Система, включающая в себя ввод данных, их электронную обработку и вывод информации, используемой для регистрации или автоматического контроля.
44 сопоставление; выход продукции (reconciliation): Сравнение ожидаемого и фактического объемов произведенной или использованной продукции с учетом стандартных отклонений.
45 спецификация (specification): Документ, содержащий требования, предъявляемые к материалам и продуктам, используемым или получаемым при производстве, являющийся основой для оценки качества лекарственных средств.
46 стерильность (sterility): Отсутствие живых микроорганизмов. Требования к проведению контроля стерильности приведены в соответствующей нормативной документации.
47 технологический процесс (production): Операции, включающие в себя приемку и обработку исходных материалов, упаковку и получение готового продукта.
48 упаковка (packaging): Все операции, в т.ч. наполнение и маркировка, проводимые с нерасфасованным продуктом для получения готового продукта.
Примечание — Стерильное наполнение, как правило, не следует рассматривать как часть процесса упаковки. В этом случае нерасфасованным продуктом считаются наполненные первичные контейнеры без окончательной упаковки.
49 упаковочный материал (packaging material): Любой материал, применяемый для упаковывания лекарственных средств, за исключением внешней упаковки, используемой для транспортирования. Упаковочные материалы делятся на первичные и вторичные в зависимости от наличия прямого контакта с продуктом.
50 уполномоченное лицо (Qualified person) : Сотрудник предприятия-производителя, принимающий окончательное решение о выпуске серии лекарственного средства,
51 чистая зона (clean area); Зона, построенная и эксплуатируемая таким образом, что в ней сведено к минимуму проникание, образование и накопление загрязнений в виде частиц и микроорганизмов.
Примечание — Типы чистых зон определены в приложении 1.
52 чистая изолированная зона (clean contained area): Зона, построенная и эксплуатируемая таким образом, что она одновременно является чистой и изолированной зоной.
53 экзотический организм (exotic organism): Биологический агент, вызывающий заболевание, отсутствующее в данной стране или географической зоне, либо являющийся объектом профилактических мер или программы по его устранению.
Сведения
о соответствии национальных стандартов Российской Федерации ссылочным международным (региональным) стандартам
Обозначения ссылочного международного стандарта
Обозначение и наименование соответствующего национального стандарта
ГОСТ Р ИСО 11137-2000 Стерилизация медицинской продукции. Требования к валидации и текущему контролю. Радиационная стерилизация
ГОСТ ИСО 14644-1-2002 Чистые помещения и связанные с ними контролируемые среды. Часть 1. Классификация чистоты воздуха
ГОСТ Р ИСО 14644-2-2001 Чистые помещения и связанные с ними контролируемые среды. Часть 2. Требования к контролю и мониторингу для подтверждения постоянного соответствия ГОСТ Р ИСО 14644-1
Национальный стандарт РФ ГОСТ Р 52249-2009 «Правила производства и контроля качества лекарственных средств» (утв. и введен в действие приказом Федерального агентства по техническому регулированию и метрологии от 20 мая 2009 г. N 159-ст)
Текст ГОСТ приводится по официальному изданию Федерального агентства по техническому регулированию и метрологии, Москва, Стандартинформ, 2009 г.
1 Подготовлен Ассоциацией инженеров по контролю микрозагрязнений (АСИНКОМ) на основе собственного аутентичного перевода Правил, указанных в пункте 4
2 Внесен Техническим комитетом по стандартизации ТК 458 «Производство и контроль качества лекарственных средств»
3 Утвержден и введен в действие Приказом Федерального агентства по техническому регулированию и метрологии от 20 мая 2009 г. N 159-ст
4 Настоящий стандарт идентичен Правилам производства лекарственных средств для человека и животных Европейского Союза (ЕС Guide to Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use) по состоянию на 31.01.2009 г., за исключением приложения 20
При применении настоящего стандарта рекомендуется использовать вместо ссылочных международных стандартов соответствующие им национальные стандарты Российской Федерации, указанные в разделе «Сведения о соответствии ссылочных международных стандартов национальным стандартам»
Источник


