Тема 8 Контроль качества лекарственных средств промышленного производства
учебно-методический материал

Учебный материал для студентов 2 курса по МДК Контроль качества лекарственных средств
Скачать:
| Вложение | Размер |
|---|---|
| tz_no_8_kk_ls_promyshlennogo_proizvodstva.docx | 36.79 КБ |
Предварительный просмотр:
Теоретическое занятие № 8
Тема : Контроль качества лекарственных средств промышленного производства.
- Структура отечественных производителей по лекарственным формам
- Контроль качества ЛС промышленного производства.
1. Структура отечественных производителей по лекарственным формам
На территории Российской Федерации осуществляют деятельность по производству лекарственных средств 460 предприятий (количество выданных лицензий на производство лекарственных средств более 730), 63 предприятия государственной формы собственности
Одной из приоритетных задач в области здравоохранения является обеспечение качества лекарственных средств, находящихся в обращении на территории Российской Федерации. При этом уровень требований, предъявляемый к лекарственным средствам, определяет уровень безопасности применения лекарственных средств на территории каждой страны.
Система государственного контроля качества лекарственных средств включает в себя:
- оценку эффективности, безопасности и утверждение стандартов качества на лекарственные средства при регистрации;
- оценку качества впервые производимых и впервые ввозимых лекарственных средств при допуске в обращение;
- экспертизу качества лекарственных средств, находящихся в обращении (выборочно);
- мониторинг качества, эффективности и безопасности лекарственных средств, находящихся в обращении;
- инспекционный контроль.
Росздравнадзором создана система выявления и изъятия из обращения недоброкачественных и фальсифицированных лекарственных средств.
Составляющими данной системы являются:
- территориальные Управления Росздравнадзора;
- испытательные лаборатории;
- единая информационная система;
- системы качества в организациях-производителях лекарственных средств, розничных и оптовых фармацевтических организациях.
Начиная с 2009 года, обеспечено бюджетное финансирование государственного выборочного контроля качества социально значимых категорий лекарственных средств, таких как:
- инсулины;
- антибиотики для внутривенного и внутримышечного введения;
- средства для наркоза;
- инфузионные растворы и растворы для парентерального питания и кровезаменители;
- фармацевтические субстанции, предназначенные для производства лекарственных средств отечественными производителями;
- цитостатики в лекарственных формах для инъекций;
препараты, получаемые из крови и плазмы донорской.
Наиболее часто встречающиеся нарушения в организации производства и контроля качества лекарственных средств:
Нарушения требований и условий, предъявляемых к организации производства и контролю качества лекарственных средств
*Описание (внешний вид)
1. Устаревшее технологическое оборудование (сколы по краям, шероховатости, излишняя пыль).
2. Использование упаковки, не обеспечивающей защиту препарата от внешних воздействий (свет, влажность), или отсутствие вторичной упаковки (механические повреждения).
3. Изучение стабильности лекарственных средств в условиях естественного и стрессового старения (в различных режимах температуры и влажности) осуществляется не должным образом.
4. Несоблюдение технологических стадий производства, нарушение регламентных норм, приводящих к несоответствию продукции при хранении и транспортировке (поверхность таблеток, покрытых оболочкой, с нарушенной целостностью покрытия, сколы).
*Примеси (выше установленных норм)
1. Отсутствие контроля качества в полном объеме
2. Отсутствие аудита поставщиков (производителей) активных фармацевтических ингредиентов и, как следствие, выбор поставщиков сырья по принципу закупок «ЦЕНА-качество», вместо «КАЧЕСТВО-цена».
3. Привлечение к контролю качества контрактных лабораторий, которые не владеют спецификой анализа конкретных препаратов или методиками проведения анализа, как следствие, получение недостоверных результатов испытаний.
4. Нерепрезентативный отбор проб на всех стадиях контроля производственного цикла.
1. Недостаточная отработка технологии производства генерических препаратов (приготовление и сушка гранулята, режим работы пресс-инструментов):
— в случае изменения состава вспомогательных веществ;
— в случае введения технологических «ноу-хау» оригинальных препаратов.
1. Несоответствие санитарного состояния и класса чистоты производственных помещений видам проводимых в них работ.
2. Отсутствие санитарного мониторинга производственной среды, отсутствие микробиологических лабораторий на производстве, проведение микробиологического контроля на контрактной основе не в полном объеме требований.
3. Несоответствие качества исходного сырья и материалов.
1. Несоответствие классов чистоты в производственных помещениях видам работ, проводимых в них.
2. Использование первичной упаковки (ампулы, флаконы, пробки, крышки) низкого качества.
3. Отсутствие современного оборудования для контроля стерильных растворов на механические включения.
4. Водоподготовка и контроль воды очищенной и для инъекций не отвечают современным требованиям.
- Контроль качества ЛС промышленного производства.
Министерство здравоохранения Министерство экономики
Российской Федерации Российской Федерации
от 03.12.1999 № 432/512
«О введении в действие Стандарта отрасли ОСТ 42-510-98
«Правила организации производства и контроля качества лекарственных средств
Стандарт отрасли ОСТ 42-510-98
- Представляет собой свод правил по организации производства и контроля качества лекарственных средств;
- Стандарт распространяется на все предприятия, выпускающие готовые лекарственные средства, независимо от ведомственной подчиненности и форм собственности.
В России построены предприятия с производственными помещениями по GMP (более 15 площадок), однако система обеспечения качества ЛС на большинстве предприятий слаба или отсутствует. Подтверждение тому – «милдронатовый» кризис, использование в производстве фальсифицированных субстанций и др.
Каковы системные проблемы российской фармацевтической промышленности?
— отсутствие утвержденной национальной концепции развития фармацевтической промышленности (в настоящее время она разработана);
— низкий уровень инноваций и технологий, используемых при разработке и производстве лекарственных средств (высшим руководством страны поставлена задача повысить этот уровень);
— отсутствие механизмов финансирования разработок лекарственных препаратов;
— низкий уровень российского патентного законодательства и законоприменительной практики относительно международных стандартов;
— непрерывно истощающийся кадровый потенциал отечественной науки и производства;
— сырьевая зависимость отечественной промышленности.
Существует острая потребность в подготовке квалифицированных специалистов для современных фармацевтических производств, обладающих знаниями по новейшим технологиям производства лекарств (в основном ГЛС-таблетирование, капсулирование, ампулирование), современному оборудованию, материалам, а также действующим стандартам производств (GMP и др.) со знанием современных норм и правил по регистрации и сертификации. Проблемой является не столько отсутствие кадров как таковых (формально обучение проводится в большом количестве учебных заведений), сколько дефицит подготовленных специалистов мирового уровня. Мы практически не имеем специалистов, владеющих вопросами контроля качества, способных работать в условиях GMP. В настоящее время лицензию на подготовку кадров по специальности «Фармация» имеют 48 вузов и только 2 вуза (в Курске и С.-Петербурге) выпускают инженеров химико-технологического профиля и специалистов для фармацевтических производств.
Первый – чрезвычайно важный! – шаг на пути к переходу российской фармпромышленности на стандарты GMP сделан: принят Федеральный закон от 12.04.2010 № 61-ФЗ «Об обращении лекарственных средств». Сейчас предстоит принять подзаконный нормативный акт, обязывающий предприятия выполнять требования GMP (закон обязывает предприятия перейти на стандарты GMP к 2014 г.).
Одновременно с этим необходимо развитие собственного производства не только готовых лекарственных форм, но и сырья. Чтобы снизить зависимость России от импортных субстанций, необходимы значительные инвестиции в модернизацию производства, в т.ч. закупку оборудования, финансирование научных разработок, создание маркетинговой стратегии реализации субстанций и др.
В основе концепции GMP лежит принципиально новый подход к обеспечению качества лекарственных средств, а именно переход от контроля качества готовой продукции к обеспечению ее качества во время процесса производства. При этом объектом контроля в первую очередь становятся сам процесс производства и различные производственные факторы (здания, помещения, оборудование, персонал и т.д.). Поэтому только соблюдение принципов, требований и норм правил GMP на фармацевтических предприятиях гарантирует выпуск эффективных и безопасных лекарственных средств надлежащего качества.
Особое значение в деятельности работников фармацевтической отрасли имеют требования GSP — надлежащей практики хранения лекарственных средств.
Хранение и транспортировка фармацевтических материалов и продукции имеет место на всех этапах их обращения, и в этих операциях задействованы практически все участники фармрынка. Поэтому нельзя с уверенностью говорить о качестве, безопасности и эффективности лекарственных средств, не разработав национального или хотя бы внутрифирменного стандарта хранения лекарственных средств, соответствующего нормам GSP, и не соблюдая его на практике.
Высокий (надлежащий) уровень качества должен поддерживаться в сфере обращения фармпродукции таким образом, чтобы допущенные к продаже лекарственные средства реализовывались оптовой и розничной торговлей без изменения их свойств..
Правила GDP устанавливают требования по отношению к персоналу, документальному оформлению (заказов, операций, отчетов), помещениям и оборудованию, поставкам клиентам, возврату продукции (доброкачественной и бракованной), самоинспекциям и т.д.
Важной с точки зрения качественного хранения является дата повторного испытания (retest date), когда материал должен быть вновь исследован, чтобы гарантировать его пригодность для использования. В России датой переконтроля считается дата повторного анализа, указанная на упаковке дата, вплоть до которой лекарственная субстанция, при условии надлежащего обращения и хранения, может использоваться в качестве исходного материала для производства лекарственных продуктов без проверки соответствия требованиям фармакопейной или иной спецификации. После этой даты субстанция может использоваться по назначению лишь при условии получения положительного результата по итогам полного анализа (испытания) на соответствие спецификации. В этом случае устанавливается новая дата повторного анализа.
Так, чтобы качественно и гарантированно достичь целей хранения фармпродукции, на каждом участке хранения (будь то производитель, дистрибьютор, оптовик, аптека или ЛПУ) должно быть адекватное количество квалифицированных сотрудников. Весь персонал должен пройти надлежащее обучение Правилам GSP, выполнять соответствующие инструкции, процедуры и меры безопасности. Все сотрудники организации должны соблюдать высокий уровень личной гигиены и санитарии. Персонал, работающий в зонах хранения, должен носить подходящую защитную рабочую одежду, соответствующую виду деятельности, которую он выполняет.
Образцы должны приниматься только соответственно обученным и квалифицированным персоналом в строгом соответствии с инструкциями по осуществлению выборки, если это требуется. Упаковки, из которых отобраны образцы, должны быть соответственно маркированы. После осуществления выборки товар должен быть изолирован. Изоляция партии должна поддерживаться в течение карантина и последующего хранения. Материалы и фармпродукция должны оставаться на карантине до тех пор, пока не будет полученное уполномоченное подтверждение или отклонение. Должны быть приняты меры, чтобы гарантировать, что забракованные материалы и фармпродукция не могут использоваться. Они должны храниться отдельно от остальных материалов и продуктов до ожидаемой ликвидации или возвращения поставщику [1] .
Источник
КОНТРОЛЬ КАЧЕСТВА ЛЕКАРСТВЕННЫХ СРЕДСТВ ПРОМЫШЛЕННОГО ПРОИЗВОДСТВА
Производство ЛС — это серийное получение ЛС в соответствии с правилами организации производства и контроля их качества, утвержденными соответствующим федеральным органом.
Производство ЛС осуществляется предприятиями-производителями, имеющими лицензии на их производство. Государственный контроль производства выполняют федеральный и территориальный органы контроля качества ЛС, в права которых входит:
— беспрепятственный доступ на любое предприятие-производитель ЛС и контрольное изъятие производимых образцов;
— снятие копий с документов, необходимых для проведения контроля производства и качества ЛС;
— запрет производства и продажи уже произведенных ЛС в случаях: 1) если ЛС не прошли государственную регистрацию в РФ (исключение — ЛС, предназначенные для проведения клинических исследований); 2) отсутствия лицензии на производство; 3) изготовления с нарушением правил организации производства и контроля качества ЛС (статья 13 Государственного Закона о лекарственных средствах).
Лицензия на производство ЛС выдается федеральным органом исполнительной власти на срок не менее 5 лет. В случае, если предприятием-производителем изменены условия производства, оно обязано получить новую лицензию на производство.
На фармацевтическом предприятии контроль качества ЛС является частью надлежащей производственной практики, гарантирующей правильность процедуры отбора проб, подготовки пробы к анализу (пробоподготовки), определения действующего вещества, принятия решения о приеме испытуемого материала в соответствии с требованиями, установленными в НД (ФСП, спецификации).
Отдел контроля качества (ОКК) фармацевтического предприятия осуществляет различные типы фармацевтического контроля: входной контроль фармацевтических субстанций и вспомогательных веществ, межоперационный контроль в процессе производства и контроль качества готовой продукции (рис. 6.2).
Таким образом, контроль качества на производстве включает контроль исходного сырья, полупродуктов, лекарственных субстанций и готовых лекарственных форм.
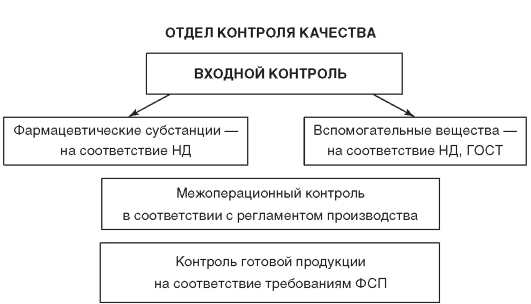
Рис. 6.2. Типы фармацевтического анализа в условиях производства лекарственных средств
(по:Елизарова Т.Е. Современные методы стандартизации и контроля качества лекарственных средств. — М.: МИА, 2008)
В период бурного развития фармацевтической промышленности возникли проблемы качества ЛС, которые не могли быть решены только путем усиления фармакопейного анализа. Обеспечение качества ЛС стало возможным только на базе проведения фармацевтического анализа в соответствии с правилами GMP.
Согласно правилам GMP, объектом контроля становится весь процесс производства ЛС, включая помещения, персонал, документацию. В России правила GMP нашли отражение в виде ГОСТ Р 52249-2009 «Правила производства и контроля качества лекарственных средств».
Стандарт содержит требования к производству и контролю качества ЛС для человека и животных. Стандарт распространяется на все виды ЛС и устанавливает общие требования к их производству и контролю качества, а также специальные требования к производству отдельных видов ЛС. Стандарт не распространяется на обеспечение промышленной безопасности, пожарной безопасности, взрывобезопасности, химической безопасности и безопасности других видов при производстве ЛС, требования к которым приведены в других НД.
Надлежащая практика контроля качества фармацевтических препаратов обеспечивается комплексом мероприятий при их разработке и исследовании с учетом требований GMP. Каждая методика
должна содержать обоснование преимуществ по сравнению с другими в виде представленных результатов сопоставления ее применения (валидация).
Валидация методавключает следующие метрологические характеристики:
— правильность (accuracy) — близость результатов к истинному значению, что может быть проведено при сравнении с результатами, полученными с помощью иной методики, валидированной ранее;
— точность (precision) — согласованность между отдельными результатами испытаний (отклонение отдельных результатов от среднего значения — относительное стандартное отклонение);
— сходимость (repeatability) — точность методики при ее выполнении одним и тем же аналитиком при одних и тех же условиях (реактивы, оборудование, лаборатория);
— воспроизводимость (reproducibility) — точность методики при использовании ее в различных условиях для идентичных образцов, отобранных из одной и той же однородной серии материала (разные лаборатории, исполнители, оборудование, время);
— надежность (robustness) — способность методики давать результаты анализа с приемлемой правильностью и точностью при изменении условий работы для предположительно идентичных образцов из одной и той же однородной серии материала;
— чувствительность (sensitivity) — способность методики испытания регистрировать небольшие изменения концентрации (наклон калибровочной кривой);
— предел обнаружения (limit of detection) — наименьшее содержание, при котором анализируемое вещество может быть обнаружено.
Контроль качества ЛС на отдельных технологических стадиях его получения обеспечивает надлежащее качество конечного продукта.
Для лекарственных препаратов регламентируется надлежащая микробиологическая чистота. Загрязнение микроорганизмами может происходить на разных стадиях производства, поэтому испытания на микробиологическую чистоту проводят на всех стадиях получения ЛС. Основными источниками микробной контаминации являются сырье, вода, оборудование, воздух производственных помещений, упаковка готовой продукции, персонал.
Для количественного определения содержания микроорганизмов в воздухе используют различные методы отбора проб: фильтрацию, осаждение в жидкостях, осаждение на твердые среды. Для оценки микробиологической чистоты выполняют тесты на стерильность.
При определении стерильности ЛС, обладающих выраженным антибактериальным действием, бактериостатическими, фунгистатическими свойствами, а также содержащих консерванты, используют метод мембранной фильтрации.
Метод мембранной фильтрации основан на пропускании ЛС через полимерную мембрану. При этом микроорганизмы остаются на поверхности мембраны. Затем мембрану помещают в соответствующую питательную среду и наблюдают образование колоний при инкубировании.
Также проводят испытания на пирогенность инъекционных препаратов.Необходимость проведения этого теста связана с присутствием в инъекционных препаратах фрагментов клеток грамположительных и грамотрицательных бактерий, грибов, вирусов и эндотоксинов. Применение таких препаратов вызывает жар, озноб, тошноту, иногда летальный исход. Наибольшую опасность представляют эндотоксины, которые являются термостабильными и состоят из липополисахаридов внешней плазматической мембраны грамотрицательных бактерий, попадание которых в организм человека провоцирует резкий воспалительный процесс.
Контроль содержания пирогенных примесей в инъекционных препаратах проводят двумя методами: в опытах in vivo на кроликах и in vitro, с использованием ЛАЛ-реактивов, приготовленных из водного экстракта (лизата) амебоцитов мечехвоста полифема 1 .
ЛАЛ-тест впервые был включен в Фармакопею США в 1980 г., а позже был признан и в европейских странах. В 1997 г. в РФ была утверждена ФС «Определение содержания бактериальных эндотоксинов (ЛАЛ-тест)».
Основными преимуществами ЛАЛ-теста по сравнению с традиционным испытанием на кроликах является возможность оценки уровня бактериальных эндотоксинов в тех препаратах, которые нельзя испытывать на животных; более высокая (в 100 раз) чувствительность ЛАЛ-теста; быстрота выполнения (одно испытание занимает около 1,5 ч); испытания проводит один человек.
Более высокая чувствительность и быстрота ЛАЛ-теста обеспечивают возможность контроля качества воды для приготовления инъекционных ЛС в условиях производства. Этот тест используется для анализа «Воды инъекционной в ампулах», «Раствора натрия хлорида 0,9% для инъекций», различных лекарственных форм инсулина.
Под чувствительностью ЛАЛ-реактива понимают минимальную концентрацию международного.стандарта эндотоксина, которая приводит к образованию плотного геля при реакции с данным ЛАЛ-реактивом в гель-тромб тесте, проведенном при стандартных условиях.
Испытания на кроликах проводят на 12 кроликах породы шиншилла массой 2,5-3,0 кг с соблюдением строгого стандартного рациона питания в специально оборудованном тихом, светлом, без перепадов температуры помещении. Кроликов помещают в определенным образом оборудованные индивидуальные боксы, позволяющие проводить постоянное измерение температуры, которая записывается в автоматическом режиме самописцем, что исключает фальсификацию данных.
В начале опыта препарат вводят 3 животным. ЛС считается апирогенным, если суммарное повышение температуры тела животных (∑Δt) составляет не более 1,2 °С. На втором этапе испытаний препарат вводят 6 животным. ЛС признают пирогенным, если ∑Δt>3,0 °С. На третьем этапе число животных равно 9. ЛС считают пирогенным при ∑Δt>4,5 °С. Наконец, на четвертом этапе используется 12 животных, и препарат считают пирогенным, если ∑Δt>5,4 °С. После окончания проверки составляют протокол, который включает параметры эксперимента, заключение о соответствии проверяемого раствора требованиям на пирогенность.
Метод имеет ряд недостатков. В эксперименте используют животных, чувствительность которых к пирогенам в 3-4 раза ниже, чем у человека. Это требует соответствующего увеличения тест-дозы. Кроме того, многие ЛВ в дозах, близких к терапевтическим, могут вызвать токсические реакции и даже гибель животных,
поэтому используется заниженная величина тест-доз (инфузионные растворы глюкозы, антибиотики — бензилпенициллина натриевая соль, линкомицина гидрохлорид и др.). На проведение одной серии опыта требуется около 5 ч.
1 Мечехвост полифем, краб-подкова, королевский краб. Вид: Limulus polyphemus (Linnaeus, 1758).
LAL(ЛАЛ)-тест (Limulus Amebocyte Lysate) — ферментативная реакция ЛАЛреактива с эндотоксином, в результате которой образуется плотный гель. При отсутствии эндотоксина в анализируемой пробе гель не образуется. Положительный результат ЛАЛ-теста указывает на то, что в исследуемой пробе содержание эндотоксина составляет не менее 0,25 ЕЭ (единиц эндотоксина). Отрицательный результат говорит о том, что в исследуемой пробе содержание эндотоксина — менее
Контрольные вопросы и задания
• Охарактеризуйте этапы фармацевтического анализа: определение подлинности, оценка чистоты (определение примесей), количественный анализ.
• Перечислите правила надлежащей деятельности (GP), в соответствии с которыми должны осуществляться производство и контроль качества ЛС.
• Каковы особенности фармацевтического анализа воспроизведенных ЛС?
• Перечислите типы эквивалентности ЛС. Как производят оценку фармацевтической и биологической эквивалентности?
• Объясните необходимость применения в фармацевтическом анализе стандартных образцов сравнения.
• Перечислите характеристики валидации аналитического метода.
Источник


