Биодоступность




![]()
![]()
БД определяется как относительное количество лекарственного вещества, достигшее системного кровотока и скорость, с которой этот процесс происходит. Относительное количество вещества, т.к. степень БД определяется в сравнении исследуемойлекарственной формы и стандартной.При этом используют одинаковые дозы стандартной и исследуемой лекарственной формы. СБД выражают в %.:
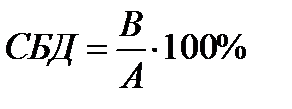
где А — количество лекарственного вещества, всосавшегося в организм после назначения стандартной лекарственной формы; В — количество лекарственного вещества, всосавшегося в организм после назначения исследуемой лекарственной формы.
Различают абсолютнуюБД, при этом в качестве стандартной лекарственной формы при определении применяют раствор для внутривенного введения. При этом способе введения вся доза лекарственного вещества поступает в большой круг кровообращения.
На практике чаще определяют относительнуюБД. В этом случае стандартом является хорошо всасываемая для данного способа применения лекарственная форма, например, раствор или суспензия для пероральных лекарственных форм (таблеток, гранул); раствор или суспензия в виде мйкроклизмы для ректальных лекарственных форм (суппозиториев).
БД определяют на живых организмах, т.е. в опытах «in vivo»,— на животных при проведении доклинических испытаний, на людях — добровольцах при клинических испытаниях. Известны две группы методов определения БД: фармакодинамические и фармакокинетические.
Фармакодинамические— основаны на измерении эффектов, вызываемых лекарственным веществом, или биохимических реакций на лекарственное вещество или его активные метаболиты. Например, фиксируется реакция зрачка, изменение сердечного ритма, изменения болевых ощущений или биохимических показателей после введения лекарственного препарата.
Более объективны и менее сложны фармакокинетическиеметоды, основанные на измерении уровня концентрации лекарственного вещества в крови в зависимости от времени, или его метаболитов в моче.
При фармакокинетических методах определения БД проводят последовательный забор проб крови, мочи и др. биожидкостей в течение определенного времени после введения препарата в пробах чувствительными аналитическими методами определяют концентрацию лекарственного вещества.
Разработаны более простые методы «in vitro»(в пробирке), позволяющие косвенно определить БД по скорости и степени высвобождения лекарственного вещества из лекарственной формы, или методы, моделирующие всасывание лекарственного вещества «in vitro».
При методах «in vitro» термин БД заменяют на термин «фармацевтическая доступность»(ФД).
Для определения фармацевтической доступности предложено множество методов и приборов.
Однокамерные приборы со статическими условиями растворения и с применением средств перемешивания, например, для определения фармацевтической доступности лекарственного вещества в таблетках, гранулах, драже, капсулах с твёрдым содержимым, используют тест «Растворение» с использованием приборов «вращающаяся корзинка» и «лопастная мешалка» (см. ОФС «Растворение»),
Для оценки фармацевтической доступности лекарственных веществ в мягких лекарственных формах используют методы, основанные на диффузии лекарственного вещества из лекарственной формы:
1) диализные методы (через мембраны);
2) метод прямой диффузии в различные среды: агаровый, коллагеновый гели.
Источник
Полиморфизм: влияние на качество лекарственных средств и актуальные методы анализа
Полный текст:
Аннотация
Ключевые слова
Для цитирования:
Гильдеева Г.Н. Полиморфизм: влияние на качество лекарственных средств и актуальные методы анализа. Качественная Клиническая Практика. 2017;(1):56-60.
For citation:
Gildeeva G.N. Polymorphism: the influence on the quality of drugs and actual methods of analysis. Kachestvennaya Klinicheskaya Praktika = Good Clinical Practice. 2017;(1):56-60. (In Russ.)
Введение
Полиморфизм во многом определяет свойства веществ. Во многих отраслях науки и техники это свойство вещества активно исследуется. Полиморфные модификации (ПМ) образуют многие химические, в том числе и лекарственные, вещества. Полиморфизм объясняется тем, что одни и те же атомы вещества могут образовывать различные устойчивые кристаллические решётки [2, 3].
Образование различных полиморфов одного и того же лекарственного вещества обычно происходит при изменении условий кристаллизации, при введении в жидкие или мягкие лекарственные формы различных вспомогательных веществ, при сушке и т.д. Учёт и рациональное использование явления полиморфизма веществ имеют большое значение для медицинской практики. Практически от того, какая кристаллическая модификация субстанций содержится в лекарственном препарате, зависит его стабильность и эффективность [4].
По этой причине лекарственные субстанции, полученные на разных заводах-изготовителях, иногда даже в пределах одной серии на одном и том же за-воде-изготовителе, могут различаться по физико-химическим свойствам, что определяется особенностями технологии их получения, особенно на этапе кристаллизации, а также возможностью полиморфных переходов при транспортировке и хранении. Такие переходы возможны как при производстве готовых лекарственных средств, так и при их хранении, что может влиять на свойства готовой продукции.
В фармации явление полиморфизма стало изучаться сравнительно недавно в рамках биофармацевтического направления. В нормативной документации на лекарственную субстанцию обычно не отмечается факт возможного наличия у него полиморфизма и не оговаривается необходимость соблюдения определённых условий кристаллизации вещества, обеспечивающих получение кристаллов с наиболее высокой терапевтической эффективностью, хотя в некоторых случаях в нормативной документации на лекарственное вещество, например, на рокситромицин, отмечается способность вещества к полиморфизму, но при этом не предлагается проведение испытаний по вопросу идентификации полиморфных форм и не описываются способы их получения [5].
Всё вышесказанное обуславливает необходимость изучения полиморфизма лекарственных веществ и разработки алгоритма поиска наиболее активных их полиморфов и методов их анализа.
Полиморфизм как характеристика, определяющая ключевые свойства активной фармацевтической субстанции
Полиморфизм активной фармацевтической субстанции (АФС) может быть причиной фармацевтической неэквивалентности и, как следствие, фармакокинетической и терапевтической неэквивалентности препаратов в твердой лекарственной форме. Имеющиеся существенные отличия в растворимости полиморфных модификаций АФС могут привести к различию кинетики их растворения in vivo и, как следствие, — биодоступности лекарственных средств (ЛС).
Биодоступность/биоэквивалентность ЛС зависит от ряда факторов, определяющих скорость и степень их абсорбции, таких как кинетика растворения и проникающая способность через мембраны клеток, гастроинтестинальная подвижность, метаболизм. Эти факторы учитываются в концепции биофармацевтической классификационной системы (БКС) АФС, которая уже принята в руководствах регулирующих органов в промышленно развитых странах [6].
Большинство литературных данных укладываются в рамки БСК АФС и позволяют предсказать на основании фармацевтических свойств, будет ли различаться биодоступность ПМ АФС. Тем не менее встречаются и противоречивые сведения относительно биодоступности кристаллических модификаций лекарственных веществ (ЛВ) [7]. Возможными причинами противоречий, по-видимому, являются следующие:
- растворимость ПМ АФС часто сравнивается в апротонных органических растворителях или в деионизированной воде, т.е. в условиях отличных от процесса абсорбции АФС;
- в подобных работах не учитывается степень дисперсности отдельных ПМ АФС.
В контексте исследования биофармацевтических свойств АФС в литературе практически не рассматривается вопрос, чем обусловлен наблюдаемый фармакокинетический эффект конформационных ПМ АФС: лишь разницей в растворимости и скорости растворения отдельных модификаций, либо отчасти и сохранением конформационного отличия ПМ АФС в водном растворе?
Согласно Европейской Фармакопее, свойства ПМ АФС в растворах и расплавах идентичны. Между тем первые наблюдения, указывающие на отличие свойств растворов ПМ низкомолекулярных органических соединений, были отмечены в первой половине ХХ в. Относительно недавно было сообщение о том, что раствор метастабильной ПМ местного анестетика дикаин проявляет в 2—3 раза более положительную анестезирующую активность по сравнению со стабильной модификацией при местном применении на роговицу глаза [8, 9], при субарахноидальном введении препарата, как средства спинальной анестезии. При этом токсичность дикаина в 3 раза превышает токсичность метастабильной ПМ. Эта группа авторов отмечает и более высокую репаративнорегенеративную активность бета-ПМ метилурацила по сравнению с его альфа-модификацией на клетки переднего эпителия роговицы глаз, что может объясняться различной диффузией через мембрану двух бионеэквивалентных форм метилу- рацила. Учитывая низкобарьерный характер конформационных состояний ПМ, сохранение различий между ПМ АФС в растворе после диссолюции объяснялось за счёт их стабилизации кооперативными взаимодействиями, в частности, за счёт внутримолекулярных связей, образования молекулярных ассоциатов и сольватации [10]. Действительно, супрамолекулярные ассоциаты возникают на стадии образования зародышей при кристаллизации [11]. Однако в условиях, отличных от пересыщенных растворов, молекулы АФС сольватированы. Например, результаты исследования флуоресценции дифлунизала в растворе и в четырёх кристаллических ПМ не позволяют предположить возможность кооперативных взаимодействий в растворе [12].
В настоящее время большое внимание учёных уделяется хиральности как фундаментального свойства биологических систем [13]. Так, например, до последнего времени амлодипин использовали в виде рацемической смеси право- и левовращающих изомеров. Вместе с тем установлено, что способность блокировать кальциевые каналы L-типа принадлежит преимущественно левовращающему S-энантиомеру [14]. Изучение амлодипина показало, что присоединение к дигидропиридиновым рецепторам является стерео-селективным, и связь с S(—) изомером была в 1000 раз сильнее, чем с R(+)изомером [15]. Стереоселективность рецепторов к S(—) и R(+)изомерам объясняет различия в клиренсе, биодоступности и клинической активности препарата. Применение чистого левовращающего фармакологически активного S(—)изомера амлодипина вместо рацемической смеси имеет важные преимущества, ведь необходимая доза и системная токсичность могут быть снижены.
По-видимому, значимые различия в свойствах растворов ПМ можно ожидать для АФС большой молекулярной массы — липофильных, обладающих конформационной жёсткостью полициклических соединений, в которых дополнительно реализуются сильные внутримолекулярные взаимодействия, например, стероидов, гормонов, пептидов, природных высокомолекулярных биологически активных веществ, характеризующихся высокой пространственной организацией молекул [16]. Однако этот вопрос в полной мере не может быть решён без систематического исследования конформаций молекул ПМ АФС в растворах методами спектроскопии циркулярного дихроизма и объёмного ядерного магнитного резонанса.
С учётом вышесказанного важным вопросом является стабильность кристаллических модификаций АФС. Метастабильная ПМ может превращаться в термодинамически более стабильную форму, что в свою очередь может привести к уменьшению растворимости АФС. Высокая дисперсность АФС может негативно влиять на стабильность АФС при хранении за счёт агрегации [17], превращений аморфной формы, изменения полиморфного состава. В процессе хранения вне плотной упаковки, в замороженном состоянии возможны кристаллизация аморфных образцов и образование гидратов, превращение метастабильных полиморфных модификаций в стабильные формы [18].
Таким образом, из многочисленных факторов, которые оказывают влияние на качество твёрдых АФС, краеугольным является кристаллическая структура АФС [19, 20]. Понимание взаимосвязи кристаллической структуры, био- и фармацевтических свойств может позволить оптимизировать технологический процесс получения и состав лекарственной формы с заданными свойствами, которые должны обеспечивать оптимальную биодоступность действующего вещества. При разработке готовых лекарственных форм и аналитической нормативной документации на ЛС необходима строгая регламентация полиморфного состава и степени измельчения частиц АФС, что является принципиальным для АФС 4-го и особенно 2-го класса по БСК. В мировой практике доклинические и клинические исследования при разработке новых ЛС включают биофармацевтический скрининг, связанный с выявлением фармацевтических факторов, влияющих на высвобождение, фармакокинетику, фармакодинамику и токсикодинамику АФС.
Все аналитические методики по изучению ПМ АФС должны быть предварительно валидированы [21]. Разработан ряд руководств, в которых рассматриваются вопросы валидации аналитических процедур, например, ICH. Необходимо проверять использование описанных в фармакопеях аналитических методик для каждого состава [22]. Примером неправильного применения методики может служить состав, из которого при обработке в соответствии с фармакопейным методом образуется опалесцирующий раствор на этапе, на котором должно быть измерено поглощение УФ-излучения.
Методы анализа полиморфных модификаций лекарственных средств
Изменение кристаллической модификации ЛВ в производственном процессе, при хранении и реализации ЛС может привести к изменениям фармацевтических и, как следствие, фармакокинетических показателей и терапевтических свойств ЛС. В связи с этим идентификация и количественное определение ПМ, сольватов и аморфной формы ЛВ имеет ключевое значение при характеристике ЛВ в процессе разработки и производства ЛС, а также для контроля качества АФС и ЛП. Данная практика действует в странах Международной конференции по гармонизации (ICH), где соответствующие испытания являются обязательными для регистрации новых ЛС. В большинстве стран и, в частности, в России для регистрации новых ЛС исследование полиморфизма ЛВ не является обязательным.
Руководство ICHQ6A [23], определяющее требования к качеству новых АФС и ЛП, рекомендует аналитические процедуры испытания полиморфизма и условия принятия критериев качества ЛС. Согласно данному руководству, для идентификации и характеристики ПМ ЛВ рекомендуются следующие методы:
- Дифракция рентгеновских лучей, являющаяся прямым методом идентификации и анализа ПМ и определения степени кристалличности ЛВ.
- Спектральные методы, позволяющие выявить различия между внутримолекулярными взаимодействиями ПМ ЛВ.
- Термоаналитические методы для исследования фазовых переходов в ходе нагревания/ охлаждения и для количественной характеристики процессов десольвации ЛС;
- Оптическая и сканирующая электронная микроскопия для исследования морфологии кристаллов ЛС.
Что касается положения в России, то в настоящее время для контроля качества ЛС применяют достаточно большое количество методов. Соответствующие методики включают в нормативную документацию (НД): фармакопейную статью предприятия (ФСП) на отечественные лекарственные средства и НД — на зарубежные.
Как показывают наши исследования и данные других авторов, для оценки качества и выявления фальсификатов лекарственных средств, особенно фармацевтических субстанций, уже недостаточно применения стандартных аналитических методов. И, в частности, лишнее тому подтверждение — явление полиморфизма.
Действительно, можно провести установление подлинности стандартными фармакопейными методами — ИК, УФ, хроматография, химические реакции. Это позволит, например, отбраковать поддельные субстанции. Можно провести анализ чистоты теми же стандартными подходами (хроматография, УФ-спектрофотометрия, анализ примесей). Это тоже важно, в том числе и с точки зрения выявления фальсификатов. Можно провести количественное определение, и это также позволит отбраковать ряд лекарственных средств.
Но остаются, например, две субстанции разных производителей, полностью удовлетворяющие всем требованиям нормативной документации по разделам «подлинность», «чистота» и «количественное определение». Однако изготовленные из этих субстанций препараты (по абсолютно одинаковой и валидированной технологической схеме) показывают различную биодоступность и стабильность.
К этому стоит добавить и тот факт, что фальсифицированные субстанции также часто не удаётся выявить по стандартной фармакопейной схеме — они удовлетворяют всем испытаниям, из-за чего ряд специалистов высказывает определённый скептицизм по отношению к сложившейся системе фармакопейного анализа.
Следовательно, возникает вопрос: какие методы анализа и какие указания следует включать дополнительно в современную НД и какие данные необходимо приводить в соответствующем регистрационном досье? Что это означает на практике, например, с точки зрения оценки полиморфизма субстанций?
Наибольшую ценность с точки зрения оценки полиморфных модификаций субстанций представляют собой рентгеновская дифракция и термоаналитические методы — дифференциальная сканирующая калориметрия и термогравиметрия.
Ценность ИК-спектроскопии для этих целей, меньше, поскольку изменения в ПМ не всегда могут приводить к регулярным изменениям в соответствующих ИК-спектрах. Этот метод стоит продолжать рассматривать как основной для установления подлинности.
Также важную информацию может дать оценка морфологии частиц субстанции методами оптической или электронной микроскопии.
Если с методами анализа всё достаточно очевидно, то есть ещё и вторая сторона вопроса — экономическая. Современный аналитический арсенал позволяет провести практически любые испытания любой степени сложности. Но стоимость соответствующего оборудования может оказаться достаточно высокой, чтобы иметь его в стандартном центре контроля качества лекарственных средств.
Руководители Центра Контроля Качества Лекарственных Средств (ЦККЛС) часто говорят о существенных финансовых затратах на оснащение лабораторий. Очевидно, что каждую лабораторию в стране невозможно оснастить по полной схеме всем парком современных приборов. И, естественно, мы тоже делим оборудование на первостепенное, которое обязательно необходимо иметь в лабораториях (в соответствии с частотой использования тех или иных методов в НД), и то, которое стоит приобретать по мере необходимости. Возможно, целесообразно использовать схему специализации, когда отдельные ЦККЛС выполняют определённые высокотехнологичные испытания, например, с использованием тех же приборов, необходимых для оценки полиморфизма продукции, поставляемой на фармацевтический рынок. Существенную роль в этом должна сыграть государственная поддержка, особенно в части оснащения лабораторий, которые могли бы работать в соответствии с требованиями сети Официальных медицинских контрольных лабораторий (OMCL).
Список литературы
1. Hilfiker R. Polymorphism in pharmaceutical industry. Weinheim: Wiley-VCH, 2006.
2. Бабилев Ф.В., Андроник И.Я. Полиморфизм лекарственных веществ. Кишинев: Штиинца. 1981; 239.
3. Смехова И.Е., Молдавер Б.Л., Громова Э.Г. Изучение влияния таутомерии на противовоспалительную активность бутаглионамида. Фармация. 1984; 6: 23-25.
4. Акашкина Л. В., Буленков Т.И., Езерский М.Л. Сравнительная оценка кристаллических форм стрептоцида. Научные труды. М.: 1983; XXI: 195-200.
5. Barton J.H. Reforming the patent system. Science. 2000; 287: 1933-1934.
6. Ahr G., Voith B., Kuhlmann J. Guidances related to bioavailability and bioequivalence: European industry perspective. Eur. J. Drug Metab. Pharmacokinet. 2000; 25: 25-27.
7. Ali A.A., Farouk A. Comparative studies on the bioavailability of ampicillin an- hydrate and trihydrate. Int. J. Pharm. 1981; 9: 3: 239-243.
8. Леонидов Н.Б., Успенская С.И., Гацуро В.В. Физико-химические свойства леокаина и особенности его биологической активности в сравнительном аспекте с дикаином. РХЖ. 1997; 16: 5: 53-60.
9. Майчук Ю.Ф., Щипанова А.И. Фармакодинамические характеристики тетракаина и его метастабильной модификации — леокаина как средств местной анестезии в офтальмологии. Росс. Хим. Журн. 1997; 16: 5: 61-63.
10. Леонидов Н.Б., Серезнев Н.Б., Успенская С.И. Исследование диффузионных свойств растворов полиморфных модификаций 6-метилурацила. РХЖ. 1997; 16: 5: 49-50.
11. Gu С.Н., Young V., Grant D.J.W. Polymorph screening: Influence of solvents on the rate of solvent-mediated polymorphic transformation. J. Pharm. Sci. 2001; 90: 1878-1890.
12. Brittain H.G. et al. Solid-state fluorescence studies of some polymorphs of diflunisal. J. I. Pharm. Res. 2005; 22: 6: 999-1006.
13. Решетова Е.Н., Горбунов А.А., Аснин Л.Д. Препаративное хроматографическое разделение энантиомеров ибупрофена на хиральной неподвижной фазе Whelk 01. Химико-фармацевтический журнал. 2012; 9: 47-51.
14. Лутай М.И., Лысенко А.Ф., Моисеенко О.И. Использование оптических изомеров известных сердечно-сосудистых средств — путь к повышению их эффективности и переносимости. Укр. кардіол. журн. 2009; 4: 13-20.
15. Арсеньева К.Е. Применение амлодипина в кардиологической практике. РМЖ. 2009; 17 (8): 610-613.
16. Gurjar M. The future lies in chiral purity: A perspective. J. Indian Med. Assoc. 2007; 105 (4): 177-178.
17. Rabinow В.Е. Nanosuspensions in drug delivery. Nature Rev. 2004; 3: 9: 785-796.
18. Glass B.D., Novak Cs., Brown M.E. The thermal and photostability of solid pharmaceuticals: A review J. Therm. Anal. Cal. 2004; II: 1013-1036.
19. Lennard M.S. Clinical pharmacology through the looking glass: reflections on the racemate vs enantiomer debate. Br. J. Clin. Pharmac. 1991; 31: 623-625.
20. Lien A.N., He H., Pham-Huy C. Chiral Drugs: An Overview. International Journal of Biomedical science. 2006; 2: 2: 85-100.
21. Srinivas N., Barbhaiya R.H., Midha K.K. Enantiomeric drug development: issues, considerations and reculatory requirements. J. Pharmacol. Sci. 2000; 90: 1205-1215.
22. Харитонов Ю.Я. Аналитическая химия (аналитика): в 2 кн. М.: Высшая школа, 2001; 615.
23. ICH Guidance Q6A specification: Spetifications of new drug substances and products: Chemical substances. 1999.
Источник


