1.Всасывание лекарств. Пресистемная элиминация. Пролекарства. Сравнительная биодоступность при сублингвальном, оральном и ректальном способах введения.
Пути введения ЛС: а. энтеральный путь введения: перорально, сублингвально, трансбуккально, ректально, через зонд; б. парентеральный путь введения: внутривенно, подкожно, внутримышечно, ингаляционно, субарахноидально, трансдермально.
Основные механизмы всасывания:
1.Пассивная диффузия через мембрану клеток. Определяется градиентом концентрации веществ.Чем выще липофильность вещества, тем легче оно всасывается через клеточную мембрану.
2. фильтрация через поры мембран. Диаметр пор в кишечнике не велик , поэтому через них диффундирует вода, некоторые оины, мелкие гидорофильные молекулы.
3 Активный транспорт характеризуется избирательностью к определенным соединениям, возможна конкуренция за один транспортный механизм, насыщаемостью. Активный транспорт обеспечивет всасывание гидрофильныхтполярных молекул.
4.При пиницитозе происходит инвагинация клеточной мембраны с последующим образованием пузырька( вакусли).Пузырек мигрирует по цитоплазме к противоположной стороне клетки, где путем экзоцитоза содержимое пузырька выводится наружу.
Пресистемная элиминация лекарственных средств (эффект первого прохождения) — процесс биотрансформации лекарства до попадания лекарственых средств в системный кровоток. В пресистемной элиминации при пероральном введении лекарства участвуют ферментативные системы кишечника, крови воротной вены и гепатоциты.
Сравнительная биодоступность: сублингвально- всасывание быстрое, минуя при первом пассаже печеночный барьер и не контактируя с ферментами ЖКТ. Орально- всасывание происходит частично из желудка, главным образом в тонкой кишке. Ректально- около 50% веществ поступает в кровоток, минуя печень. Всасывание – простой диффузии.
2 Нестероидные противовоспалительные средства: характеристика группы. К ним относятся вещества, оказывающие влияние на циклооксигеназу и таким путем снижают биосинтез простаноидов. ЦОК- продуцируется в обычных условиях и регулирует образование в организме простаноидов. ЦОК-2 в значительной степени индуцируется процессом воспаления( образуется в отсутствии воспаления).
I.неизбиратеные ингибиторы ЦОК -1 и -2:
Производные салициловой кислоты( кислота ацетилсалициловая)
Производные антраниловой кислоты( кислота мефенамовая, кислота флуфенамовая)
Производные индолуксусной кислоты( индометацин)
Производные фенилуксусной кислоты( диклофенак- натрий)
Производные фенилпропионовой кислоты(ибупрофен)
Производные нафтилпропионовой кислоты( напроксен)
Оксикамы( пироксикам, лорноксикам)
II.Избирательные ингибитры ЦОК-2 ( целекоксиб, рофекоксиб).
Эффект: Жаропонижающий эффект связан с уменьшением продукции простагландина Е2.нпвс действуют только при лихорадке
Анальгезирующий эффект связан с нарушением образования Е2 и I2,которые повышают чувствительность болевых рецепторов к брадикинину.
Фармакокинетика. Все НПВС хорошо всасываются в желудочно-кишечном тракте. Практически полностью связываются с альбуминами плазмы, вытесняя при этом некоторые другие лекарственные средства. Метаболизируются НПВС в печени, выделяются через почки.
Показания. Ревматические заболевания. Ревматизм (ревматическая лихорадка), ревматоидный артрит, подагрический и псориатический артриты, анкилозирующий спондилит (болезнь Бехтерева), синдром Рейтера.
Неревматические заболевания опорно-двигательного аппарата. Остеоартроз, миозит, тендовагинит, травма (бытовая, спортивная). Нередко при этих состояниях эффективно применение местных лекарственных форм НПВС (мази, кремы, гели).
Неврологические заболевания. Невралгия, радикулит, ишиас, люмбаго
Лихорадка (как правило, при температуре тела выше 38,5°С)..
противопоказаны при эрозивно-язвенных поражениях желудочно-кишечного тракта, особенно в стадии обострения, выраженных нарушениях функции печени и почек, цитопениях, индивидуальной непереносимости, беременности.
Побочные эффекты:из-за угнетения ПГ Е2 и I2 нарушается целостность слиз оболочки жедудка и 12-ти перстной кишки.тошнота.могут появится эрозии желудка.чтобы избежать это назначают нпвс совместно с препаратами гастропротекторных ПГ.
3 НадропаринПилокарпин: полная характеристика препарата.М-холиностимулирующее средство, оказывает миотическое и противоглаукомное действие.
Вызывает сокращение циркулярной (миоз) и цилиарной мышц (спазм аккомодации), увеличивает угол передней камеры глаза (оттягивается корень радужки), повышает проницаемость трабекулярной зоны (трабекула натягивается, и происходит открытие блокированных участков шлеммова канала), улучшает отток водянистой влаги из глаза и в конечном итоге снижает внутриглазное давление
Острый приступ закрытоугольной глаукомы, вторичная глаукома, первичная открытоугольная глаукома, абсцесс роговицы; кровоизлияние в стекловидное тело. Необходимость сужения зрачка после инстилляции мидриатиков.
Гиперчувствительность, ирит, циклит, иридоциклит, кератит, состояние после офтальмологических операций и др. заболевания глаз, при которых сужения зрачка нежелательно.
Источник
Биодоступность лекарственных средств пресистемная элиминация
Морфологические барьеры организма проиллюстрированы в предыдущей статье на сайте. Физико-химические свойства лекарственного средства определяют, достигнет ли он цели, расположенной на поверхности или внутри клеток организма либо бактериальных клеток, и в какой степени.
В тех случаях, когда препарат принимается внутрь или вводится парентерально, фармакокинетические процессы идут совершенно иначе, чем при местном применении.
Это становится очевидным, если проследить путь принятого внутрь препарата, начинаяс места всасывания до попадания в кровоток. Возможен один из следующих вариантов развития событий.
1. Препарат проникает через эпителий кишечника в энтероциты, однако Р-гликопротеид транспортирует его обратно в просвет кишечника. Поэтому итоговое всосавшееся количество препарата может быть значительно меньше.
Этот «противотранспорт» различен у разных людей в отношении одного и того же вещества и, кроме того, подвержен влиянию других лекарственных средств.
2. На пути из просвета кишечника в общее циркуляторное русло принятый внутрь препарат расщепляется под воздействием ферментов, например цитохромоксидазы Р450.
(а) Разрушение может начаться уже в слизистой кишечника. Другие лекарственные средства или химические вещества могут подавлять или стимулировать активность изоферментов цитохрома в кишечнике.
Отдельный пример — сок грейпфрута, который подавляет активность оксидаз CYP3A4 в стенке кишечника и вызывает повышение в крови концентрации некоторых лекарственных средств до токсического уровня.
(б) Самую важную роль играет метаболизм в печени, которому подвергается любое лекарственное средство, попадающее в организм. В печени работает множество ферментов, химически преобразующих эндогенные и экзогенные вещества, чтобы обеспечить их выведение из организма. Только часть всосавшегося количества может попасть в кровь печеночной вены, это зависит от количества препарата, поглощенного и переработанного гепатоцитами.
Следует учитывать, что другие препараты могут вызывать повышение активности ферментов (увеличение гЭР).
Процессы, о которых говорится выше, объединены термином «пресистемная элиминация».
3. Парентеральное введение лекарственного вещества позволяет обойти пресистемную элиминацию. При в/в, п/к и в/м введении препарат проходит через полую вену, попадает в правый желудочек и, через легкие, в левый желудочек, далее — в системный кровоток.
Богатые жиром и имеющие большую поверхность легкие всасывают некоторое количество липофильных и амфифильных веществ, а затем медленно выделяют их после снижения концентрации в крови.
Во время быстрого поступления лекарственного средства в организм легкие играют роль буфера и защищают сердце от избыточной концентрации веществ после в/в введения.
В некоторых ситуациях желательна быстрая пресистемная элиминация. Яркий пример — назначение глюкокортикоидов при бронхиальной астме.
Поскольку большая часть ингалированного препарата обычно проглатывается, глюкокортикоиды с полной пресистемной элиминацией оказывают минимальную системную нагрузку на организм.
Использование клопидогрела для ингибирования агрегации тромбоцитов — пример желательной пресистемной активации.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Источник
Пресистемная элиминация
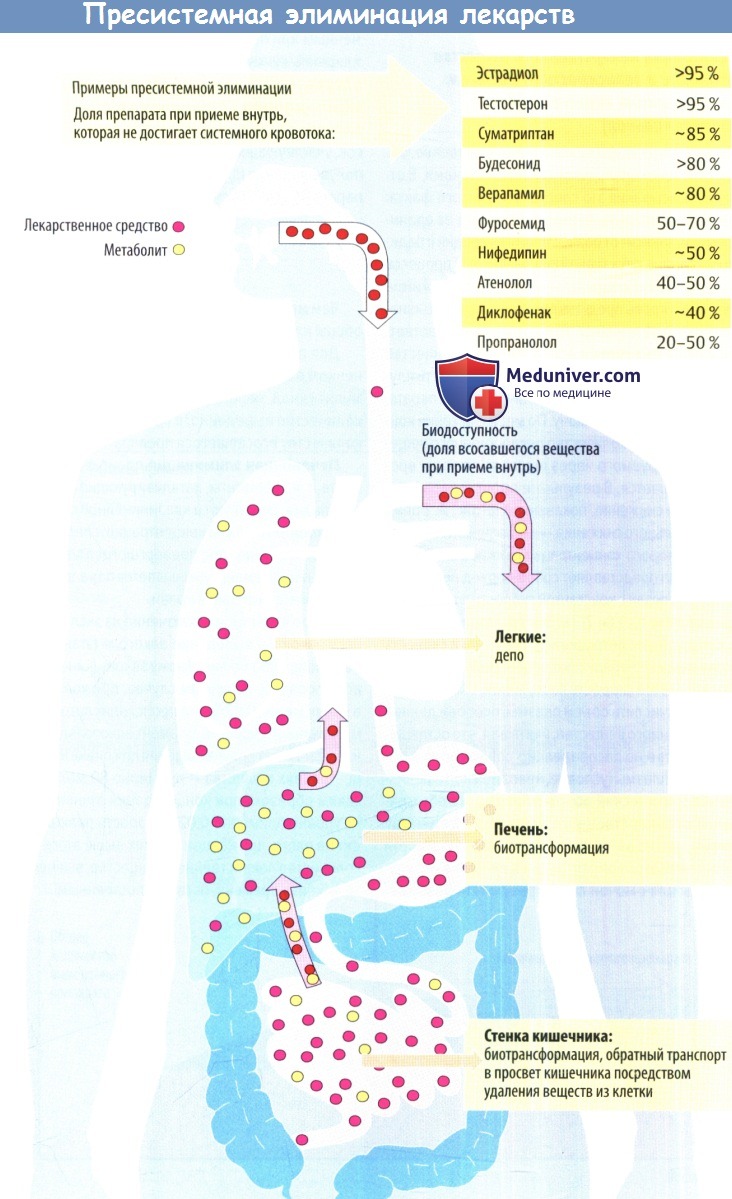
При приеме препарата внутрь, кроме биологических мембран, стоящих на пути из просвета кишечника в кровь, имеется еще один фактор, ограничивающий поступление лекарства в системный кровоток – печеночный метаболизм. Лекарственное вещество, поступая из ЖКТ по воротной вене в печень, может подвергнуться ферментативному разрушению, в связи с чем в системный кровоток попадает лишь часть (иногда незначительная часть) принятой дозы. Данный феномен носит название«эффект первого прохождения через печень». Так, некоторые лекарственные средства, обладая высокой абсорбцией, попадают в системный кровоток в очень небольшом количестве, не оказывающем терапевтического эффекта. Этот эффект характерен для быстро метаболизирующихся (см. фазу биотрансформации) средств и при значительной выраженности исключает возможность приема соответствующего препарата внутрь (например, антиаритмик лидокаин). В других случаях его можно корригировать увеличением дозы, которая оказывается значительно выше, чем при внутривенном введении (верапамил, морфин, пропранолол). Весьма демонстративным в этом плане является пример нитроглицерина. Эффект первого прохождения через печень у этого препарата достигает 85-97% дозы, что объясняет необходимость его назначения «в обход» печени (сублингвально или внутривенно) и делает бессмысленным нередко встречающееся назначение внутрь «капель Вотчала» (раствор нитроглицерина в ментоле).
Часть введенной дозы, достигшая системного кровотока, является важнейшей характеристикой препарата. Последняя обозначается как «биодоступность» и по определению ВОЗ понимается как степень и скорость, с которой вещество или его активная часть доставляется из лекарственной формы в системный кровоток. При внутривенном введении биодоступность принимается за 100%. При приеме внутрь она зависит от ряда факторов: устойчивости лекарства к действию соляной кислоты желудочного сока, активности разрушения препарата ферментами в просвете и стенке кишечника, выраженности эффекта первого прохождения через печень, то есть от потерь вследствие, так называемой,пресистемной элиминации.
Пресистемная элиминация зависит не только от препарата, но и от ряда факторов организма пациента и условий применения лекарства. На пресистемную элиминацию влияют взаимодействия с пищей и другими лекарственными средствами (см. гл. 9), скорость эвакуации из желудка, моторная функция кишечника, состояние функции печени и портального кровообращения. При нарушении функции печени (при циррозе), а также при развитии системы анастомозов между воротной веной и полыми венами (при портальной гипертензии) лекарственное средство попадает в системный кровоток, минуя печень. При этом снижается эффект первого прохождения через печень, что может вести к передозировке лекарства несмотря на назначение терапевтической дозы.
Кроме перечисленных факторов на биодоступность влияют еще и особенности технологии приготовления лекарственной формы. Поэтому биодоступность одного и того же активного вещества, выпускаемого в разных лекарственных формах или в лекарствах различных производителей, может колебаться в широких пределах. Однако такие колебания существенно затрудняют эффективное и безопасное дозирование лекарственных средств. В связи с этим перед регистрацией лекарства необходимо провести исследование биодоступности нового препарата в сравнении с эталонным лекарственным средством. В результате получается информация о сравнительной биодоступности или о биоэквивалентности.
Тут вы можете оставить комментарий к выбранному абзацу или сообщить об ошибке.
Источник
Биодоступность пероральных препаратов
*Импакт фактор за 2018 г. по данным РИНЦ
Читайте в новом номере
В условиях интенсивного развития фармацевтической промышленности и огромного разнообразия препаратов для перорального приема у специалистов возникает необходимость обновлять свои знания о препаратах этой категории и процессах, происходящих в организме при их всасывании.
Современные фармацевтические технологии позволяют изменять в определенном диапазоне фармакокинетические параметры перорально принимаемого лекарственного средства. Как правило, эти технологии направлены на повышение биодоступности лекарственного средства и/или уменьшение риска возникновения нежелательных реакций. Объективной характеристикой количества всосавшегося вещества является площадь под кривой концентрация–время (AUC).
На основные фармакокинетические параметры перорально принятого препарата (максимальная концентрация, время ее достижения, период полувыведения, константа скорости элиминации и др.), кроме его физико–химических свойств, существенное влияние могут оказывать состояние желудочно–кишечного тракта пациента и физиологические процессы в системе пищеварения.
В связи с этим представляется важным рассмотреть факторы, влияющие в организме человека на биодоступность лекарственной формы при пероральном приеме.
Физиологические процессы в ЖКТ, влияющие на биодоступность
пероральных лекарственных форм
При пероральном приеме активное вещество таблетки (пока она не растворилась) проходит последовательно ротовую полость, пищевод, желудок, тонкий кишечник.
В ротовой полости таблетка обволакивается слюной. Многие лекарственные формы для перорального приема покрыты специальной оболочкой, препятствующей воздействию на них ферментов слюны, поэтому препараты, назначаемые перорально, не рекомендуется разжевывать.
Длина тонкой кишки – 5 м (двенадцатиперстной – 27–30 см). Пища находится в желудке от 30 мин. до полутора часов, в тонкой кишке – около 4 часов. Как правило, те же самые временные промежутки сохраняются и для лекарственных препаратов, принятых через рот.
Процесс усвоения некоторых лекарственных веществ начинается уже в желудке. Играет роль не только кислотность желудочного сока, но и время опорожнения желудка. У больных с высокой кислотностью желудочного сока вследствие спазма пилорического отдела замедляется опорожнение желудка, в результате чего всасывание лекарственных средств также замедляется. При анацидном состоянии опорожнение желудка наступает быстро, и это приводит к ускорению всасывания лекарственных средств и более быстрому наступлению фармакодинамического эффекта.
Из желудка лекарственное средство поступает в двенадцатиперстную кишку, куда открывается общий желчный проток и проток поджелудочной железы. Компоненты желчи способствуют растворению липофильных препаратов, оболочек, капсул, таблеток с кишечнорастворимым покрытием. В кишечнике активное вещество высвобождается из лекарственной формы и взаимодействует с кишечным соком. При этом соли желчных кислот могут образовывать с некоторыми лекарственными средствами нерастворимые комплексы, что приводит к снижению их биодоступности.
Большинство перорально принимаемых веществ всасывается в тонком кишечнике, имеющем чрезвычайно развитую поверхность (около 200 м2). Скорость поступления в системный кровоток при этом зависит от кровоснабжения кишечника в зоне всасывания.
На процесс всасывания лекарственных веществ существенное влияние оказывает пища. Для большинства лекарственных средств характерно замедление всасывания под влиянием пищи, связанное с замедлением опорожнения желудка. Особенно замедляет эвакуацию желудочного содержимого горячая, кислая, жирная, чрезмерно соленая или сладкая пища, а также пища густой консистенции. Но в некоторых случаях длительное пребывание лекарственных средств в желудке, способствует их более полному растворению и после перехода химуса в тонкую кишку биодоступность может повыситься (например, нитрофурантоин, гипотиазид). В связи с этим прием лекарственных препаратов связывают с режимом питания [1].
Во–первых, пища может выступать в качестве механического барьера, препятствующего контакту лекарственного средства с эпителием кишечника. Во–вторых, ряд продуктов могут оказывать влияние на рН содержимого желудка. В–третьих, пища может взаимодействовать с лекарственными средствами с образованием хелатных комплексов.
Препарат рекомендуется принимать до еды, если нужно быстро создать высокую концентрацию в крови. В остальных случаях считается целесообразным назначать лекарственные препараты после еды. Лекарственные средства, характеризующиеся значительной биотрансформацией при первом прохождении через печень, целесообразно принимать сразу после еды, при этом их биодоступность увеличивается за счет уменьшения пресистемной элиминации.
Следует отметить, что снижение биодоступности при приеме с пищей некоторых лекарственных препаратов не считают показанием к их назначению перед едой, так как при этом лекарственное средство может оказать раздражающее действие, вызвать обострение гастрита, язвенной болезни и способствовать развитию диспептических явлений.
Учитывая особенности фармакокинетики витаминов, их целесообразно принимать во время еды.
Энтеральный (пероральный) путь введения лекарственного средства является самым распространенным в практической медицине.
Он наиболее удобен и относительно безопасен для пациента. Однако для самого препарата это наиболее долгий и трудный путь, в результате которого происходят естественные потери самого активного вещества, достигающего рецепторного аппарата. В связи с этим некоторые вещества не имеют лекарственной формы для приема внутрь, так как они разрушаются ферментами желудочно–кишечного тракта (например, инсулин и другие белки), кислой средой желудка (например, бензилпенициллин).
Механизмы всасывания
Самый простой механизм транспорта лекарственных веществ – пассивная диффузия через мембраны клеток кишечной стенки (энтероцитов). Скорость всасывания в этом случае пропорциональна градиенту концентрации веществ и существенно зависит от их растворимости в мембране (наиболее легко таким путем всасываются липофильные неполярные вещества). Диффузии, как правило, подвергаются электролиты, находящиеся в недиссоциированном состоянии. Растворимость и степень ионизации лекарственного средства определяются рН содержимого желудка и кишечника. Необходимо подчеркнуть, что лекарственные средства путем пассивной диффузии хорошо всасываются не только в тонкой, но и толстой, и прямой кишках, что служит основой для разработки многих лекарственных средств с замедленным выделением действующего вещества, а также введения лекарственных средств ректальным путем.
Вода, электролиты и малые гидрофильные молекулы (например, мочевина) транспортируются в кровь другим механизмом – фильтрацией через поры в эпителии кишечника.
Активный транспорт, использующий специализированные механизмы клеточных мембран и молекулы–переносчики, обеспечивает всасывание гидрофильных полярных молекул, неорганических ионов, аминокислот, пиримидинов. Он требует для своего осуществления затрат энергии и характеризуется избирательностью, насыщаемостью, возможностью транспорта против градиента концентрации. При активном транспорте часто наблюдается конкуренция веществ за общий транспортный механизм (например, при усвоении некоторых витаминов и минеральных веществ). Степень всасывания зависит от дозы препарата, так как возможен феномен «насыщения белков–переносчиков».
Основной механизм всасывания ксенобиотков (синтезированных) лекарственных веществ – пассивная диффузия, активный транспорт используется редко. Для веществ природного происхождения, таких как аминокислоты, витамины, эссенциальные микроэлементы и др., в организме имеются специализированные активные транспортные механизмы. В этом случае основной путь усвоения – активный транспорт, а пассивная диффузия начинает играть роль только при очень высоких концентрациях.
Лекарственные вещества с большими молекулами или комплексы лекарственного вещества с крупной транспортной молекулой всасываются путем пиноцитоза. При этом происходит инвагинация мембраны клетки кишечного эпителия и образование пузырька (вакуоли), заполненного захваченной жидкостью вместе с лекарством. Вакуоль мигрирует по цитоплазме клетки к противоположной стороне и освобождает содержимое во внутреннюю среду организма. Однако пиноцитоз не имеет существенного значения для всасывания лекарственных средств и используется лишь в редких случаях (например, при усвоении комплекса цианокобаламина с белком – внутренним фактором Кастла) [1,2].
Фильтрация через поры имеет значение для всасывания лекарственных средств с молекулярной массой менее 100 Да.
Современные технологии
управляемого высвобождения
в производстве лекарственных средств
Современные аналитические методы позволяют определять в плазме крови сверхнизкие концентрации исследуемых лекарственных веществ, что дает возможность строить фармакокинетическую кривую с большой точностью и, соответственно, с большей определенностью судить о ее параметрах. Это в сочетании со знанием механизма усвоения конкретного вещества при пероральном приеме позволяет целенаправленно разрабатывать его лекарственную форму.
Для пероральных таблетированных препаратов применяются такие технологические приемы, как:
– использование вспомогательных веществ;
– гранулирование;
– микрокапсулирование;
– применение специального прессования;
– покрытие оболочками и т.д.
С их помощью можно изменять время распада таблетки, скорость растворения или выделения лекарственного вещества, место выделения и длительность нахождения в определенной зоне желудочно–кишечного тракта (над окном всасывания). А это, в свою очередь, определяет скорость и полноту всасывания, динамику концентрации лекарственного вещества в крови, то есть биодоступность препарата [3].
К сожалению, большинство применяемых в фармацевтике технологий производства таблетированных препаратов не позволяют независимо влиять на время и на место усвоения активного вещества, так как обычно препарат непрерывно продвигается по желудочно–кишечному тракту вместе с пищевым комком или химусом. То есть задержка времени высвобождения активного вещества неизбежно сдвигает место высвобождения ниже по пищеварительному тракту. Для некоторых конкретных препаратов предлагаются оригинальные методы решения этой проблемы: таблетки из микрочастиц с адгезивными свойствами, которые «приклеиваются» к слизистой оболочке, или таблетки, разбухающие в желудке настолько, что плавают на поверхности и/или не могут пройти через пилорический сфинктер в кишечник [4].
На скорость распада таблетки в желудке влияет способ их производства. Так, обычные (прессованные) таблетки прочнее тритурационных (формованных). Скорость распада зависит и от вспомогательных веществ, используемых для придания необходимых свойств таблетируемой смеси (сыпучесть, пластичность, прессуемость, содержание влаги и т.д.).
Кишечнорастворимые таблетки получают путем покрытия их желудочно–резистентной оболочкой или прессованием гранул или микрокапсул, предварительно покрытых такими оболочками. При необходимости оболочки могут обеспечивать и более длительную задержку растворения, чем на 1 час, который таблетка проводит в желудке.
Оболочка может быть достаточно толстой, например, сахарной, которая иногда имеет большую массу, чем ядро таблетки, содержащее лекарственное вещество. Тонкие пленочные оболочки (менее 10% от массы таблетки) могут выполняться из целлюлозы, полиэтиленгликолей, желатина, гуммиарабика и т.д.
Подбором оболочки и введением дополнительных веществ можно достичь замедления нарастания концентрации активного вещества в крови, что важно для снижения риска развития нежелательной реакции и/или сдвинуть время достижения максимума на несколько часов, если требуется продлить действие препарата и тем самым сократить кратность приема в целях повышения комплаентности.
Таблетки пролонгированного действия (ретард), например, обычно получают прессованием микрогранул лекарственного вещества в биополимерной оболочке или распределеннием в биополимерной матрице. При постепенном (послойном) растворении основы или оболочки высвобождаются очередные порции лекарственного вещества.
Современные высокотехнологичные способы доставки позволяют достичь постепенного равномерного высвобождения лекарственного вещества. Например, за счет создания осмотического давления внутри капсулы с действующим веществом. На этом принципе созданы новые лекарственные формы известных препаратов нифедипина (Procardia XL, Pfeizer), оксибутина хлорида (Ditrophan XL, Ortho–McNeil), метилфенидата (Concerta, ALZA) [5,6].
Управляемое (контролируемое) высвобождение может достигаться использованием в таблетках микрокапсул с лекарственным веществом, покрытых специальным полимером. После растворения внешнего слоя внутрь капсулы начинает поступать жидкость и по мере растворения ядра происходит постепенное высвобождение и диффузия лекарственного вещества через мембрану капсулы [7].
Основным фактором, ограничивающим производство и использование подобных лекарственных форм, остается условие необходимости высвобождения всего действующего начала за время прохождения таблеткой основных мест всасывания лекарственных средств в желудочно–кишечном тракте – 4–5 часов.
Проблемы использования технологий управляемого высвобождения для производства комбинированных
препаратов
Особые технологические проблемы ставят перед разработчиками комбинированные препараты, содержащие несколько активных веществ, требующих для оптимального всасывания различных условий.
Разумеется, если требования к месту и времени усвоения для компонентов одинаковы, можно просто таблетировать смесь или при необходимости (например, для ограничения контакта между компонентами при хранении) предварительно гранулировать и капсулировать компоненты.
Если компонентам требуются различные отделы ЖКТ для оптимального всасывания (желудок и тонкий кишечник или проксимальные и дистальный отделы тонкого кишечника), то таблетки прессуют из гранул с разными скоростями растворения. В этом случае возможно также использование технологий многослойного таблетирования или контролируемого высвобождения (с несколькими компартментами).
Если компоненты комплексного препарата должны усваиваться в разное время (но в одном месте желудочно–кишечного тракта), то альтернативы раздельному приему нет. Примером могут служить некоторые пероральные контрацептивы.
Обычно в состав комбинированного лекарственного средства не включают компоненты, отрицательно влияющие на сохранность, усвоение или фармакологическое действие друг друга. С витаминно–минеральными комплексами дело обстоит гораздо сложнее. Многие из них содержат в одной таблетке десятки компонентов, между которыми возможны описанные антагонистические взаимодействия. Закономерны следующие вопросы. Насколько целесообразно объединение в одной таблетке такого большого количества биологически активных веществ? Могут ли современные фармацевтические технологии создать такую лекарственную форму, которая обеспечила бы оптимальное всасывание всех компонентов при одновременном приеме?
Особенности всасывания витаминов
и микроэлементов
Рассмотрим особенности всасывания витаминов в желудочно–кишечном тракте (табл. 1).
Все витамины подразделяются на два класса в зависимости от их растворимости: жирорастворимые (липофильные) и водорастворимые (гидрофильные). К первым относятся витамины A, D, E и K, ко вторым – все витамины группы B, витамины С и H (биотин). Естественно, что растворимость существенно влияет на всасывание.
Жирорастворимые витамины могут перейти в водную среду лишь в составе мицелл, образующихся при эмульгировании желчью (солями желчных кислот) жиров в проксимальном отделе тонкого кишечника. Там же происходит всасывание этих витаминов, т.е. их освобождение из мицелл внутрь клеток кишечной стенки (энтероцитов), транспорт особыми гликопротеинами (хиломикронами) из цитоплазмы энтероцитов в лимфу и кровь. Всасывание жирорастворимых витаминов происходит в основном путем пассивной диффузии и зависит от наличия жиров в химусе.
При всасывании водорастворимых витаминов пассивная диффузия играет заметную роль только при приеме нагрузочных (высоких) доз. При приеме витаминных комплексов, содержащих компоненты в профилактических дозах, основное значение имеет активный транспорт. Механизм транспорта различен для разных витаминов.
В состав профилактических витаминно–минеральных комплексов наиболее часто включают в виде солей следующие макро– и микроэлементы: кальций, магний, железо, медь, йод, селен, цинк, марганец.
Как и витамины, эти минералы всасываются в основном в тонком кишечнике. Для активного транспорта во внутреннюю среду большинству из них требуются переносчики. Однако специфичность транспортного процесса не так велика, как в случае витаминов. Поэтому для минералов нередка конкуренция за общий транспортный механизм, когда присутствие в кишечнике одного минерала снижает всасывание другого. Так, в присутствии кальция и магния усвоение железа может снизиться на 50%.
Минералы могут снижать всасывание и некоторых витаминов, влияя на их растворимость или нарушая работу специфических механизмов активного транспорта. Так, ионы кальция и магния уменьшают растворимость витамина в присутствии меди, цинка или железа снижается всасывание рибофлавина.
Известны также примеры межвитаминного взаимодействия, когда один витамин инактивирует другой или нарушает его всасывание.
Так, витамин С окисляет кобаламин уже в таблетке и блокирует его всасывание при растворении таблетки в пищеварительном тракте.
Для эссенциальных микронутриентов, входящих в состав комбинированных витаминно–минеральных препаратов, известны десятки подобных негативных взаимодействий.
К сожалению, рассмотренные выше современные лекарственные формы выпуска витаминно–минеральных комплексов могут предотвратить только часть таких нежелательных взаимодействий.
Наиболее просто предотвратить нежелательный контакт компонентов в период хранения. Например, раздельное гранулирование смесей, содержащих витамины С и В12, позволяет предохранить последний от окисления.
Но если требуется учесть несколько подобных взаимодействий, то усложнение технологического процесса оказывается неприемлемым по экономическим соображениям.
Нежелательных взаимодействий микронутриентов в желудочно–кишечном тракте, когда компоненты–антагонисты имеют разные места всасывания, можно избежать, если использовать при таблетировании отличающиеся по времени растворения гранулы или делать многослойные таблетки. К сожалению, большинство микронутриентов наилучшим образом усваиваются в одной и той же зоне желудочно–кишечного тракта – в проксимальном отделе тонкого кишечника, который химус проходит за достаточно короткое время (около 3 ч).
Например, для того чтобы предотвратить снижение усвоения железа из таблетки витаминно–минерального комплекса, предлагалось помещать железо в труднорастворимое ядро таблетки, а кальций и другие двухвалентные металлы вводить в растворимый внешний слой [14]. К сожалению, метод оказался неэффективным, так как к моменту высвобождения и растворения ядра таблетка покидала оптимальное для всасывания в ЖКТ место.
Практически невозможно технологическими приемами снизить эффект негативных взаимодействий витаминно–минерального комплекса на метаболических путях организма. Для этого требуется согласованное изменение фармакокинетики компонентов [15].
Максимально эффективным методом предотвращения негативных взаимодействий между компонентами витаминно–минеральных комплексов на сегодняшний день является разделение приема микронутриентов–антагонистов по времени с интервалом в 4 часа.
Источник


