Анализ вэжх лекарственных препаратов

Составной частью комплексного лечения ожогов пищевода является местная терапия, которая должна проводиться в соответствии с фазами развития патологического процесса. Сотрудниками кафедр фармацевтической технологии и детской хирургии АГМУ для местной терапии химических ожогов пищевода разработаны и запатентованы мукоадгезивные композиции на основе производного целлюлозы в форме геля для перорального приема. С целью увеличения срока годности и возможности промышленного производства составы получены в форме «сухих суспензий» – однородных смесей лекарственных веществ со стабилизаторами, к которым добавляют необходимое количество растворителя непосредственно перед применением. Препараты получили условные названия «Премелтоп» (применение в фазу воспаления) и «Ремелтоп» (применение в фазу грануляций) [1,3].
При подготовке нормативной документации (ФСП, регламенты) на экспериментальный препарат «Ремелтоп» была разработана методика анализа лекарственных веществ, входящих в состав препарата, методом ВЭЖХ.
Цель данной работы – проведение валидационных исследований ВЭЖХ-методики качественного и количественного анализа экспериментального препарата «Ремелтоп».
Материалы и методы исследования
Объект исследования – порошок для приготовления суспензий для приема внутрь: «Ремелтоп» по 7,5 г (в 100,0 готовой к применению суспензии: метилурацила (ФС 42-0256-07) 2,0; метронидазола (ФС 42-0257-07) 0,75; регенкура (ФС 42-3395-97) 4,0; ароматизатора, идентичного натуральному 0,5, натрия сахарината (ФС 42-1826-82) Е 954 0,24) для применения в фазу грануляций и формирования рубца.
Экспериментальные исследования проведены на жидкостном хроматографе LC-20 фирмы «SHIMADZU» (Япония) с УФ-детектором, с последующей компьютерной обработкой результатов с использованием программы «LCsolution version 1.4». Неподвижная фаза – хроматографическая колонка 4,6×150 мм PerfectChrom C-18, размер частиц 5 нм («MZ-Analysentechnik», Германия). Подвижная фаза – А: трифторуксусная кислота (ТФУК) раствор 0,1 %, Б: ацетонитрил 100 %. Температура колонки – 35 °С, скорость подачи элюента – 100 мкл/мин, объем пробы – 20 мкл, градиентное элюирование – изменение концентрации элюента Б от 10 % до 50 %, продолжительность анализа – 8 мин.
Методика. Около 0,05 (точная навеска) порошка помещали в мерную колбу вместимостью 25 мл и обрабатывали смесью ацетонитрил-вода в соотношении (1 : 9), перемешивали в течение 5 минут, доводили объем раствора до метки тем же растворителем. Центрифугировали в течение 10 минут и фильтровали через бумажный фильтр. Полученный фильтрат вводили в колонку хроматографа как анализируемую пробу. Для приготовления элюентов использовали ацетонитрил для хроматографии «Сорт 1» («Криохром», Россия), ТФУК марки «ч.д.а» («ВЕКТРОН») и воду очищенную, полученную на установке для получения воды аналитического качества «УПВА-5».
Результаты исследования и их обсуждение
Согласно современным рекомендациям ICH и ведущих фармакопей методика количественного определения должна быть валидирована по основным характеристикам, таким как специфичность (specificity), линейность (linearity), правильность, или истинность (accuracy, or trueness), прецизионность (precision) [5,6,7,8].
Специфичность методики. При проведении исследования установлено (рис. 1), что время удерживания метилурацила составляет 2,44 мин, метронидазола 4,03 мин, на хроматограмме пики определяемых веществ хорошо разделены между собой, пики примесей из растворителя, вспомогательных веществ и основы лекарственной формы не мешают их определению.
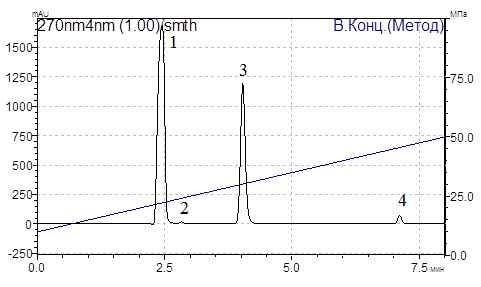
Рис. 1. Хроматограмма фильтрата состава «Ремелтоп»,
(1 – метилурацил (2,44 мин), 3 – метронидазол (4,03 мин), 2,4 – вспомогательные вещества (2,82 и 7,10 мин))
Из данных, представленных в табл. 1, видно, что спектральные отношения анализируемых веществ идентичны соответствующим спектральным отношениям стандарта. Времена удерживания метилурацила и метронидазола не существенно отличается от времени удерживания соответствующих веществ стандарта и не превышают нормы, указанной в технической документации прибора (0,5 %).
Времена удерживания и спектральные отношения компонентов состава «Ремелтоп»
Источник
Анализ вэжх лекарственных препаратов
Лечение сифилитической инфекции по-прежнему представляет проблему в связи со значительным числом неудач терапии и развития состояния серорезистентности. Фармакокинетический мониторинг мог бы помочь в решении данной проблемы. Первоначальным этапом внедрения в клиническую практику является сложный и трудоемкий процесс валидации ВЭЖХ-методики применительно к конкретной аналитической задаче. Валидация ВЭЖХ-методик, направленная на оценку качества лекарственного препарата, регламентирована специальными разделами в фармакопеях Российской Федерации (XIII изд.), Европейского союза и США, протоколы валидации подробно представлены в рекомендациях Международной Вашингтонской конференции по гармонизации (ICH) 1994 года и в специальном руководстве FDA. Универсальных протоколов валидации методик фармакокинетических исследований в настоящее время не существует. Такой протокол должен составляться каждый раз в зависимости от поставленной задачи, а основные валидационные характеристики должны критически рассматриваться применительно к используемой биологической матрице. Для внедрения фармакокинетического мониторинга терапии сифилиса необходимо решение двух основных аналитических задач. Первая – определение в крови и цереброспинальной жидкости низких концентраций препарата. Область линейности при определении бензилпенициллина натриевой соли (БПНС) методом ВЭЖХ, установленная в нескольких аналитических методиках, составляет 1–20 мкг/мл [1]. За минимальную трепонемоцидную концентрацию (МТК) принят уровень БПНС, равный 0,018 мкг/мл [2]. Таким образом, МТК находится в диапазоне низких концентраций, что затрудняет оценку терапевтической эффективности антибиотикотерапии. Вторая – решение проблем определения β-лактамов, связанных с низкой экстракцией полярных веществ из биологического материала и относительно низкими коэффициентами молярного поглощения в УФ-диапазоне (215–230 нм) [3].
Целью настоящей работы являлась разработка и апробация методологии процесса валидации при внедрении ВЭЖХ-методики определения пенициллинов и цефалоспоринов в плазме крови и цереброспинальной жидкости в процессе терапии сифилиса.
Материалы и методы исследования. Научно-информационный поиск в российских и зарубежных электронных базах данных, анализ литературы и проверка на практике всех этапов процесса валидации. Использовалась система Smartline (Knauer), состоящая из насоса Pump 1050, термостата колонки ColumnOver 4050, диодно-матричного детектора PDA Detector 2800, который позволяет снимать весь спектр в процессе анализа на разных длинах волн, инжектора (V7452). Программное обеспечение – ClarityChromV.2.6.
Результаты исследования и их обсуждение
Подготовка ВЭЖХ-системы и выбор методики. На этапе подготовки ВЭЖХ-системы необходимо протестировать колонку, используя элюенты различной концентрации. Главные показатели качества колонки – чувствительность (амплитуда пика) и разрешающая способность колонки [4]. Мы использовали колонку Kromasil 100-5C18 (150×4,6 мм, 5 мкм). Рассчитанные параметры были удовлетворительными (отклонение не более 10–20% от заявленных производителем). При использовании колонок с высокой чувствительностью и разрешающей способностью для рутинных методик нет необходимости определять наименьшую концентрацию анализируемого вещества в образце (LOD), основным параметром, согласно требованиям Фармакопеи США, считается предел количественного определения (LOQ). При выборе методологии решался вопрос достаточной чувствительности УФ-детекции для определения минорных концентраций пенициллинов и цефалоспоринов, имеющих низкие коэффициенты молярного поглощения в УФ-области спектра. Как альтернативу рассматривали применение масс-спектрометрического детектора. Но при исследовании полярных соединений, какими являются большинство ß-лактамных антибиотиков, достаточно сложно нивелировать влияние компонентов биологической матрицы на процессы ионизации, так называемый матричный эффект. Использование ВЭЖХ с участием гидрофильных взаимодействий способствует устранению «матричного эффекта» в большей степени, чем методология обращенно-фазового режима в ВЭЖХ/МС [5]. Однако в нормально-фазовой ВЭЖХ компоненты элюентов быстро испаряются, изменяя элюирующую силу буфера, следствием чего является плохая воспроизводимость. Так как воспроизводимость результатов ВЭЖХ в биологических жидкостях является одним из самых важных показателей для фармакокинетических исследований, наш выбор остался за методом обращенно-фазовой ВЭЖХ с УФ-детектированием, в которой используют полярные растворители в качестве элюентов и модифицированные силикагели С18 для заполнения колонок. Такая система обеспечивает предсказуемость поведения любых разделяемых веществ в хроматографической системе и отличную воспроизводимость и очень широко используется при анализе лекарственных препаратов [4].
Хроматографирование проводили в изократическом режиме. При валидации ВЭЖХ-методики для определения БПНС за основу была принята аналитическая методика определения пиперациллина в плазме крови [6]. Подвижная фаза: ацетонитрил 0,1 М — водный раствор дигидрофосфата калия — изопропанол, (12:84:4 по объему), предварительно профильтрованная и дегазированная, рН 6,2. Скорость потока подвижной фазы: 0,85 мл/мин., температура 30 °С, время хроматографирования 30 мин., длина волны 215 нм, время удерживания
8 мин. При выборе ВЭЖХ-методики для определения цефтриаксона (ЦТ) мы отдали предпочтение ион-парной хроматографии, для которой можно было использовать ту же самую колонку С18, заполненную октадецилсиликагелем [7]. В качестве противоионов мы использовали тетрабутиламмония бромид. Буфер рН 7,6, детектирование при 274 нм, время удерживания
Пробоподготовка. Выбор экстракционной технологии должен соответствовать задаче исследования и пройти экспериментальную проверку на имеющейся аналитической системе. Так как мы исследуем диапазон низких значений аналита, технология пробоподготовки должна включать концентрирование экстракта биопробы. Проблема концентрирования β-лактамов достаточна серьезна из-за их термолабильности. Упаривание экстрактов после преципитации или твердофазной экстракции требует особых температурных условий (не выше 40 °С). Разработан универсальный протокол для экстракции β-лактамов пенициллинового ряда из крови с использованием SPE технологии на Purosep-200В [8], но твердофазная экстракция не всегда доступна при малом количестве биологического материала, как в случае работы с ЦСЖ.
Технология ультрафильтрации с применением мембранных фильтров типа Amicon (Millipore) была признана единственной пригодной технологией очистки от примесей в стандартном методе LC-MS, одобренном FDA, для определения содержания рифампицина и тиоридазина в биологических жидкостях [9]. Однако, в отличие от разделения лекарственных субстанций в простых водных растворах, центрифужные фильтры значительно менее пригодны для биологических образцов, так как: а) в биологических матрицах могут присутствовать ингибиторы, прочно связывающие лекарственный препарат и мешающие его фильтрации, б) полученный центрифугат не всегда совместим с подвижной фазой при хроматографировании, в) низкомолекулярные белки могут привести к технической неисправности хроматографической колонки. Мы выбрали модифицированную технологию экстракции на основе депротеинизации. Для получения более концентрированной пробы депротеинизация проводится не с избытком растворителя, как в большинстве методик, а с объемом, равным объему пробы, и после уравновешивания смеси и центрифугирования растворитель удаляется из супернатанта тем же органическим элюентом, который используется в составе подвижной фазы при хроматографировании, что приводит к увеличению концентрации антибиотика в супернатанте [10].
Доказательство специфичности. Самым первым и важным этапом валидации ВЭЖХ-методики является проверка ее специфичности. Специфичность доказана, если время удерживания на колонке (t) растворов «чистых» веществ совпадает с таковым в образцах биоматериала. При использовании диодно-матричного детектора специфичность можно также подтверждать спектральной однородностью хроматографического пика определяемого вещества, для чего разработаны специальные компьютерные программы. Если спектральная однородность хроматографического пика не доказана или время удерживания аналита в биоматериале не совпадает с таковым «чистых» субстанций, то имеет место влияние другого вещества и необходимо изменить условия хроматографирования или способ пробоподготовки. Мы подтвердили специфичность определения совпадением времени удерживания с растворами «чистых» веществ пенициллина G и цефтриаксона (Sigma): 7,9 и 5,8 минуты соответственно. В подтверждении спектральной однородности необходимости не возникло.
Построение калибровочной кривой и проверка линейности. Согласно рекомендациям ICH необходимо исследовать не менее 5 растворов с разными концентрациями в области применения методики (80–120% ожидаемого диапазона концентраций). Мы готовили стандартные растворы на основе «чистых» веществ, используя соответствующую биологическую матрицу (плазма крови, ЦСЖ). Строили калибровочную кривую методом линейной регрессии, ориентируясь на площадь пиков, при помощи программы ClarityChromV.2.6. Линейность определяется визуально по кривой регрессии. При наличии линейной зависимости определяются коэффициент корреляции, точка пересечения с осью Υ, тангенс угла наклона кривой регрессии и остаточная сумма квадратов отклонений. Допустимые пределы отклонений: для наименьшей концентрации линейного диапазона – не более 20%, для остальных концентраций линейного диапазона – не более 15%. В области низких и высоких значений чаще наблюдаются отклонения от линейности, обнаружить которые достаточно сложно. Для этого применяются два подхода: первый — построить график отклонений от линии регрессии в зависимости от концентрации или от логарифма концентрации. Равномерное распределение отклонений относительно прямой доказывает линейность методики. Другой подход состоит в анализе графика относительных ответов (данные сигнала, деленные на соответствующую концентрацию), построенного в логарифмическом масштабе [11]. Если наблюдаются отклонения от линейности по используемым критериям, то такая калибровочная кривая не может быть использована и необходимо введение внутреннего стандарта. Основные требования к внутреннему стандарту: не должен содержаться в организме, должен иметь структурное сходство с аналитом и близкие хроматографические характеристики и хорошо разделяться с основным веществом на данной ВЭЖХ-системе. Для ЦТ мы использовали в качестве внутреннего стандарта цефазолин [12], для БПНС поиск проводили самостоятельно, тестируя подобные по химической структуре и физическим свойствам соединения на ВЭЖХ-системе, лучшие результаты были получены при использовании карбенициллина динатриевой соли.
Предел количественного определения (LOQ, Limit of Quantitation). FDA, ICH рекомендованы несколько способов оценки LOQ, самый распространенный – по формуле LOQ 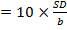 , где SD – стандартное отклонение сигнала, b – наклон калибровочной кривой. Но эта формула не учитывает требования к приемлемой воспроизводимости результатов в биологических матрицах, и есть опасность получить заниженные значения LOQ на низких концентрациях аналита. Предложен метод оценки по статистическим характеристикам калибровочного графика, который дает возможность уточнить значение LOQ, учитывая требования к воспроизводимости результатов анализа [13]. Мы применили этот удобный способ дополнительной оценки LOQ, так как можно было использовать в качестве «чистых» веществ подготовленные для калибровки растворы стандартов. LOQ важен для ответа на вопрос, возможно ли определить минимальную ингибирующую концентрацию (МИК) антибиотика с необходимой правильностью и воспроизводимостью. Если значение LOQ превышает МИК, методика неприемлема и необходимо вернуться к выбору методики, внутреннего стандарта и пробоподготовки.
, где SD – стандартное отклонение сигнала, b – наклон калибровочной кривой. Но эта формула не учитывает требования к приемлемой воспроизводимости результатов в биологических матрицах, и есть опасность получить заниженные значения LOQ на низких концентрациях аналита. Предложен метод оценки по статистическим характеристикам калибровочного графика, который дает возможность уточнить значение LOQ, учитывая требования к воспроизводимости результатов анализа [13]. Мы применили этот удобный способ дополнительной оценки LOQ, так как можно было использовать в качестве «чистых» веществ подготовленные для калибровки растворы стандартов. LOQ важен для ответа на вопрос, возможно ли определить минимальную ингибирующую концентрацию (МИК) антибиотика с необходимой правильностью и воспроизводимостью. Если значение LOQ превышает МИК, методика неприемлема и необходимо вернуться к выбору методики, внутреннего стандарта и пробоподготовки.
Проверка точности методики. ICH рекомендует проводить не менее трех определений на каждую из трех выбранных концентраций, чаще всего содержащих 80, 100 и 120% аналита от номинального значения. За номинальное значение мы приняли МТК, так как мы тестировали методику на приемлемость для работы в диапазоне низких значений, и точность определения МТК являлась наиболее важным параметром для валидации. Оценку проводили путем расчета процента нахождения с помощью тестируемой методики известного количества добавленного аналита и сравнения его с фактическими значениями введенной добавки. Процент восстановления при использовании концентраций 80, 100, 120%, скорректированный на 100%, должен был находиться в пределах 98–102% во всех сериях испытаний. Мы получали такой результат не во всех опытах с БПНС и ЦТ. Однако показано, что при работе с биоматрицами более важна воспроизводимость процента восстановления, оцениваемая по коэффициенту вариации, который не должен превышать 2% [14].
Проверка прецизионности методики. Прецизионность рассматривается на трех основных уровнях: сходимость (повторяемость), внутрилабораторная воспроизводимость и межлабораторная воспроизводимость. Сходимость – одна из характеристик надежности системы, что важно при работе с биологическими образцами. Для оценки сходимости используют коэффициент вариации (СV) в серии испытаний, включающей не менее 6 хроматограмм. Критерий приемлемости CV≤2%. Каждый из образцов готовят отдельно по выбранной методике экстрагирования и хроматографируют не менее 3 раз. Максимальный срок между испытаниями зависит от практических требований к стабильности искомых веществ. Стабильность аналита в биоматериале – важный параметр валидации ВЭЖХ-методики и требует отдельной серии испытаний и оценки [14]. Содержание β-лактамных антибиотиков при разных условиях хранения снижается на 3–18% [15]. В биоматериале при комнатной температуре и изоляции от попадания прямых солнечных лучей пенициллины стабильны в течение 3–4 часов, за это время необходимо провести все исследования. Так как большинство β-лактамных антибиотиков термолабильны, мы отказались от этапа заморозки-оттаивания.
При доказательстве внутрилабораторной воспроизводимости, то есть отсутствия систематической ошибки, используют регрессионный анализ и F-критерий Фишера [6] и показывают, что стандартные отклонения результатов испытаний, полученных в разное время, при разном порядке исследования, разными операторами, с разными реактивами статистически эквивалентны с 95% вероятностью.
Порядок оценки межлабораторной воспроизводимости приведен в Стандарте Российской Федерации ГОСТ Р ИСО 5725–2002. Требуется организовать участие не менее 5 лабораторий, оснащенных аналогичным оборудованием, что является сложным и затратным процессом. Поэтому межлабораторная воспроизводимость тестируется редко.
Заключение. На основании полученного опыта разработана схема (рисунок), в структурированном виде представляющая процесс валидации ВЭЖХ-методик для определения концентрации антибактериальных препаратов для лечения сифилиса в биологических жидкостях, в которой определены и обоснованы объем и этапность процесса и систематизированы критерии. Использование схемы при планировании и осуществлении валидационного процесса облегчит анализ и принятие решения и может быть полезно и при решении других клинических задач.
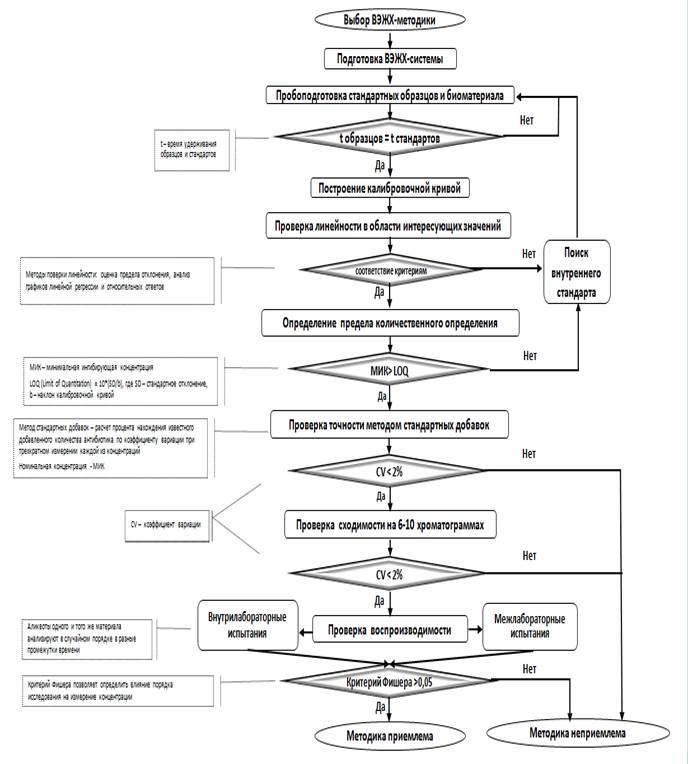
Схема валидации определения концентрации антибактериальных препаратов для лечения сифилиса в биологических жидкостях
Источник


